
Vous êtes ici
Thématique:
Conduite à tenir devant une hyperammoniémie
![]()
J. Bouchereau, S. Pichard, A. Imbard, M. Schiff*
Centre de référence maladies héréditaires du métabolisme, hôpital Robert-Debré, APHP, 48, boulevard Sérurier, 75019 Paris, France Auteur correspondant - Adresse e-mail : manuel.schiff@aphp.fr (M. Schiff).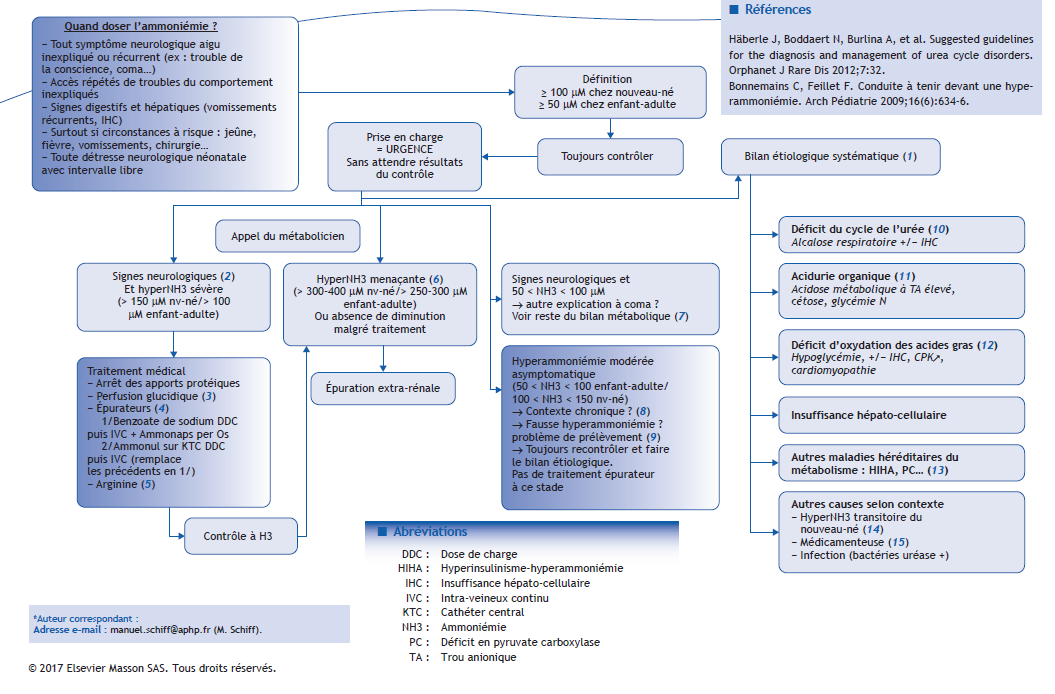
Arbre diagnostique – Commentaires
(1) Une hyperammoniémie peut être soit primitive (déficit du cycle de l’urée) soit secondaire à une autre maladie métabolique ou à des anomalies acquises. Un bilan biologique à visée étiologique doit toujours être réalisé, et comprendre :
-
gaz du sang, lactate, ionogramme sanguin, glycémie, bilan hépatique complet, hémostase, créatine phosphokinase (CPK), BU pour recherche de corps cétoniques ;
-
bilan métabolique : chromatographie d’acides aminés plasmatiques (0,5 ml tube hépariné dans la glace), profil d’acylcarnitine (peut être réalisé sur papier buvard ou plasma ; 1 ml tube hépariné dans la glace), chromatographie d’acides organiques et acide orotique urinaires (premières urines à congeler).
(2) Les signes cliniques évocateurs d’une hyperammoniémie aiguë sont ceux d’une détresse neurologique aiguë : céphalées, troubles de conscience, somnolence pouvant aller jusqu’au coma avec décès. Une hyperammoniémie peut également être évoquée devant des signes digestifs : vomissements, anorexie.
(3) Dans un premier temps, en l’absence d’orientation étiologique, les premières mesures symptomatiques sont un arrêt des apports protéiques et perfusion glucidique (soluté glucosé à 10 % contenant 5 g/L NaCl et 2 g/L KCl) apportant un débit glucidique au moins équivalent à la production hépatique de glucose, soit 8 à 10 mg/kg/min chez le nouveau-né, 5-6 mg/kg/min chez l’enfant, 4 mg/kg/min chez l’adulte. Cette perfusion permet la mise en anabolisme et l’épuration endogène de l’ammoniac et sera associée à une insulinothérapie en cas d’hyperglycémie.
(4) Les épurateurs médicamenteux permettent une épuration exogène de l’ammoniac :
-
Benzoate de sodium en première intention : dose de charge 250 mg/kg en 2 heures puis relais 250 mg/kg/j en intraveineuse (IV) continue (5,5 g/m²/j chez l’adulte, maximum 12 g/j). Ce traitement doit être débuté dès le diagnostic d’hyperammoniémie, avant transfert vers une unité spécialisée ou une réanimation.
-
Phénylbutyrate de sodium (Ammonaps®) si suspicion de déficit du cycle de l’urée, en association avec le benzoate : si voie entérale possible, 250 mg/kg/j en fractionné ou en entéral continu (5,5 g/m²/j chez l’adulte, maximum 12 g/j).
-
N-carbamylglutamate (Carbaglu®), utile dans les déficits en NAGS (N-acétylglutamate synthase) et dans les aciduries organiques : par voie entérale, 100 mg/kg en dose de charge puis 25-62,5 mg/kg toutes les 6 heures.
-
Sodium benzoate/Sodium phénylacétate (Ammonul®) en 2e intention (remplace benzoate de sodium + Ammonaps® : en IV sur voie centrale, dose de charge 250 mg/kg en 2 heures puis relais 250 à 400 mg/kg/j en IV continue (5,5 g/m²/j chez l’adulte, maximum 12 g/j). L’Ammonul® pourra être débuté d’emblée, dès que le produit est disponible (sur autorisation temporaire d’utilisation [ATU]) et dès qu’une voie centrale est mise en place. En attendant que ces conditions soient réunies, le traitement par benzoate de sodium doit toujours être débuté dès le diagnostic d’hyperammoniémie et avant transfert vers une réanimation.
(5) L’arginine est l’un des intermédiaires du cycle de l’urée. En cas de déficit du cycle de l’urée, l’arginine devient un acide aminé essentiel. La supplémentation entérale ou intraveineuse par chlorhydrate d’arginine à la dose de 200 à 400 mg/kg/j (maximum 12 g/j) permet de relancer le cycle de l’urée. Dans certains cas, la citrulline peut également être utilisée, après réception des résultats de la chromatographie des acides aminés plasmatiques.
(6) En cas d’hyperammoniémie menaçante (> 300-400 μM chez le nouveau-né, > 250-300 μm chez l’enfant et l’adulte), le risque vital est engagé à court terme, avec un risque d’engagement par oedème cérébral. Le traitement symptomatique médicamenteux doit être débuté immédiatement avant transfert vers une unité de réanimation où sera mise en place une épuration extra-rénale. En cas d’hyperammoniémie très sévère (> 1 000 μM), la mise en place de l’épuration extra-rénale doit se discuter au cas par cas car le pronostic vital et neurologique est souvent très péjoratif.
(7) Devant une détresse neurologique aiguë sans hyperammoniémie significative, il convient de rechercher d’autres causes de coma : intoxication exogène, encéphalite… De la même façon, les hyperammoniémies modérées, secondaires à d’autres maladies héréditaires du métabolisme que les troubles du cycle de l’urée (aciduries organiques, déficits de l’oxydation mitochondriale des acides gras) peuvent s’accompagner de signes neurologiques.
(8) Une hyperammoniémie peut être découverte lors de l’investigation étiologique d’une pathologie chronique : vomissements récurrents, retard de croissance, retard de développement, autisme, troubles psychiatriques. Si l’hyperammoniémie est sévère ou menaçante lors du bilan, il convient de la prendre en charge comme décrit précédemment. Si l’hyperammoniémie est modérée (< 100 μM), il convient de la contrôler et de poursuivre le bilan étiologique.
(9) Une fausse hyperammoniémie peut survenir lorsque le prélèvement est réalisé ou acheminé dans de mauvaises conditions. Pour être fiable, le prélèvement doit être réalisé sur un tube EDTA (ethylenediaminetetraacetic acid) (au minimum 1 ml de sang), le tube doit être mis immédiatement dans la glace et acheminé rapidement au laboratoire. Prévenir le laboratoire est crucial ; un résultat doit pouvoir être obtenu en 30 minutes. Cependant, même si ce résultat paraît faux, il doit toujours être pris en compte et contrôlé rapidement. Ainsi, toute élévation de l’ammoniémie est pathologique et doit être traitée jusqu’à preuve du contraire.
(10) Le cycle de l’urée permet la détoxification de l’ammoniac issu du catabolisme des acides aminés, par transformation de l’ammoniac en urée dans le foie. Un déficit d’une des enzymes du cycle d’urée ou de l’un des transporteurs impliqués dans ce cycle se manifeste par une détresse neurologique aiguë, soit en période néonatale après un intervalle libre d’intoxication, soit plus tard à tout âge, lors d’un épisode intercurrent à l’origine d’un catabolisme (infection, jeûne prolongé, charge protéique). L’hyperammoniémie est dite primitive et souvent sévère, et le reste du bilan biologique le plus souvent normal, en dehors d’une possible atteinte hépatique avec insuffisance hépato-cellulaire (syndrome de Reye). Les causes secondaires d’hyperNH3 sont décrites dans les items (11) à (15).
(11) L’hyperammoniémie dans les aciduries organiques est souvent moins importante, et très fréquemment accompagnée d’une acidose métabolique à trou anionique plasmatique élevé, avec cétose et glycémie normale.
(12) L’hyperammoniémie des déficits d’oxydation mitochondriale des acides gras peut parfois être majeure mais s’accompagne toujours d’autres signes : hypoglycémie, myolyse, défaillance hépatique, cardiomyopathie, troubles du rythme, défaillance multiviscérale.
(13) Certaines maladies héréditaires du métabolisme peuvent s’accompagner d’une hyperammoniémie modérée et chronique, comme l’hyperinsulinisme-hyperammoniémie (HIHA), certains déficits énergétiques comme le déficit en pyruvate carboxylase (PC). Dans ces situations, il n’y a pas d’urgence à traiter cette hyperammoniémie.
(14) L’hyperammoniémie transitoire du nouveau-né est observée le plus souvent chez des prématurés. Elle serait liée à une immaturité hépatique ou à un shunt vasculaire intra-hépatique (persistance du canal d’Arantius). Le reste du bilan métabolique est toujours normal, en particulier la glutamine est normale, et cette anomalie se corrige avec l’âge.
(15) Certains traitements médicamenteux peuvent entraîner une hyperammoniémie, comme la chimiothérapie par asparaginase ou un traitement par le valproate de sodium (Dépakine®). Cependant, en cas de signes neurologiques survenant sous traitement par valproate, il convient d’éliminer une cause métabolique sous-jacente, en particulier un déficit du cycle de l’urée ou une pathologie mitochondriale.
Liens d’intérêts
Les auteurs déclarent n’avoir aucun lien d’intérêts en relation avec cet article
Références
Häberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis 2012;7:32.
Bonnemains C, Feillet F. Conduite à tenir devant une hype rammoniémie. Arch Pédiatrie 2009;16(6):634-6.