
Vous êtes ici
Thématique:
Fièvres récurrentes
B. Nevena, V. Hentgenb
aService d’immuno-hématologie et rhumatologie pédiatrique, hôpital Necker – Enfant-Malades, 149 rue de Sèvres, 75015 Paris, France bService de pédiatrie, hôpital Henri-Mignot, Versailles, France Auteur correspondant - Adresse e-mail : benedicte.neven@nck.aphp.fr (B. Neven)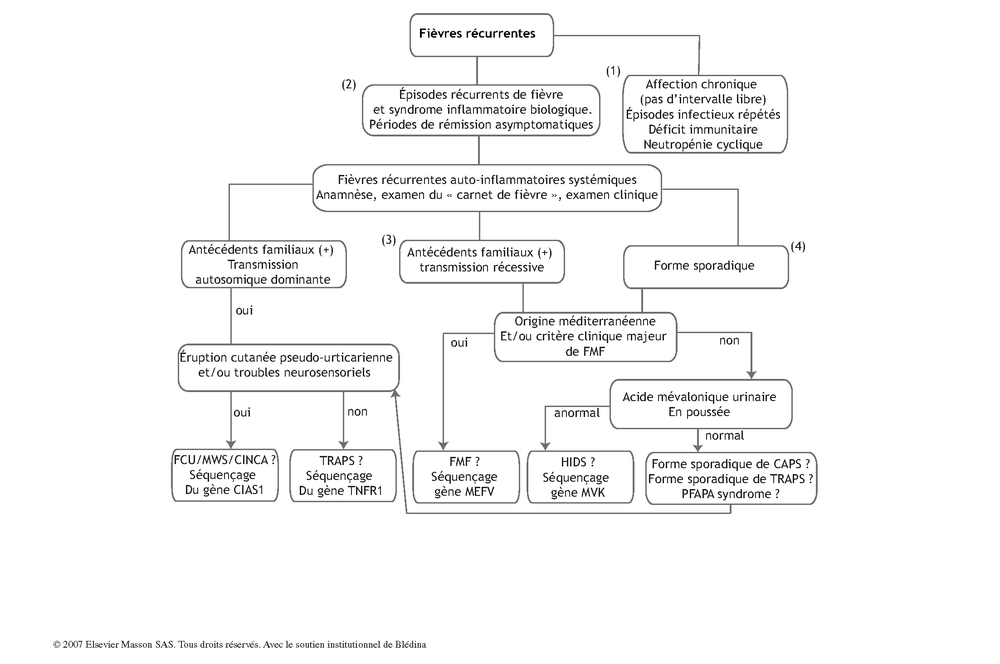
Arbre décisionnel - Commentaires
(1) Devant des épisodes de fièvres à répétition il faut :
-
s’assurer que les périodes de rémission sont parfaitement asymptomatiques. Si tel n’est pas le cas, il faut rechercher une infection chronique, une pathologie auto-immune ou une pathologie maligne ;
-
écarter des épisodes de fièvre récurrents secondaires à des épisodes infectieux répétés, éventuellement évocateurs d’un déficit immunitaire. Le plus souvent l’anamnèse suffit ;
-
écarter le diagnostic de neutropénie cyclique par l’anamnèse complétée d’une NFS en période de fièvre.
(2) Si ces causes sont écartées, il faut évoquer les syndromes auto-inflammatoires systémiques, définis par la présence d’épisodes répétés de fièvre accompagnés de signes inflammatoires d’organes (peau, articulation, séreuses…) en dehors de toute réaction immunitaire spécifique. Beaucoup de ces syndromes sont héréditaires, transmissibles selon un mode autosomique dominant ou récessif.
L’anamnèse et l’examen clinique sont essentiels pour préciser le phénotype clinique de ces patients de manière à orienter de façon adéquate le diagnostic génétique (antécédents familiaux, mode de transmission, origine ethnique, âge de survenue, fréquence des récurrences, durée des épisodes de fièvre, signes accompagnateurs). La tenue d’un « carnet de fièvre » est souvent très utile à cette étape du diagnostic clinique.
(3) Dans une minorité de cas, l’arbre généalogique est évocateur d’une pathologie héréditaire, autosomique dominante (AD) ou récessive (AR). En cas de transmission AD, il faut alors évoquer une pathologie liée au gène CIAS1 (CAPS : Cryopyrin Asssociated Periodic Syndrome) ou un TRAPS (TNF Receptor Associated Periodic Syndrome). Dans le premier cas, l’éruption pseudo-urticarienne lors des poussées de fièvre est constante. La présence de troubles neuro-sensoriels oriente également vers ce diagnostic. La fièvre méditerranéenne familiale (FMF) et le HIDS (Hyper IgD Syndrome) sont transmissibles sur un mode autosomique récessif (voir ci-dessous).
(4) La grande majorité des patients n’a pas d’antécédents familiaux particuliers. En cas d’origine méditerranéenne ou d’origine ethnique à risque, le premier diagnostic à évoquer est une fièvre méditerranéenne familiale (FMF). Après un test diagnostic et thérapeutique à la colchicine, le séquençage du gène MEFV est effectué. Si l’origine ethnique ou géographique n’est pas instructive, un dosage de l’acide mévalonique urinaire en poussée doit être effectué. S’il est anormal, le diagnostic de HIDS est hautement probable et doit être confirmé par le séquençage du gène MVK (MeValonate Kinase). Si ce dosage urinaire, effectué dans de bonnes conditions est normal, il faut évoquer une forme sporadique de CAPS ou de TRAPS, la clinique orientera les recherches génétiques. Enfin, le PFAPA syndrome est évoqué. Il s’agit de la cause la plus fréquente de fièvre récurrente sporadique de l’enfant.
Le diagnostic génétique a aussi ses limites : 20 à 40 % des patients avec un diagnostic clinique de FMF, CAPS ou HIDS n’ont pas de mutations identifiées dans le gène en cause. Le tableau ci-joint donne les caractéristiques cliniques des fièvres récurrentes appartenant aux pathologies auto-inflammatoires systémiques.
Tableau I |
|||||||
|---|---|---|---|---|---|---|---|
|
Maladie |
FMF |
HIDS |
FCAS |
MWS |
CINCA |
TRAPS |
PFAPA |
|
Mode de transmission |
AR |
AR |
AD |
AD |
AD |
AD |
Sporadique |
|
Gène |
MEFV |
MVK |
CIAS1 |
CIAS1 |
CIAS1 |
TNFRSF-1A |
|
|
Protéine |
Pyrine |
Mévalonate kinase |
Cryopyrine |
Cryopyrine |
Cryopyrine |
TNF R-15(p55) |
|
|
Durée de la fièvre |
1-3 jours |
3 à 5 jours |
1 à 2 jours |
1 à 2 jours |
variable |
7 à 21 jours |
3 à 6 jours |
|
Âge de début |
enfance |
< 1 an |
< 1 an |
< 1 an |
néonatal |
variable |
2 à 5 ans |
|
Manifestations cutanées |
+ Erythème érysipéloïde |
++ Rash maculo papuleux |
+++ Urticaire au froid |
+++ urticaire |
+++ Urticaire |
++ Eruption migrante |
|
|
Atteinte oculaire |
|
|
Conjonctivite |
Conjonctivite |
Uvéite, œdème papillaire, névrite optique |
Conjonctivite, œdème péri orbitaire |
|
|
Atteinte musculo-squelettique |
Monoarthrite ++ |
Arthralgies ++ myalgies ++ arthrites + |
Arthralgies ++ |
Arthralgies ++ arthrites ++ |
Arthralgies Arthrites ++ Arthropathies + |
Myalgies +++ Monoarthrites ++ |
Myalgies + Arthralgies + |
|
Douleurs abdominales |
+++ |
++ |
+/- |
+/- |
+/- |
++ |
|
|
Aphtose pharyngite |
|
++ ++ |
|
|
|
|
++ ++ |
|
Adénopathies |
|
++ |
|
|
+ |
+ |
++ |
|
Amyloidose |
+ |
rare |
|
+ |
+ |
+ |
|
|
Test diagnostic, Signes spécifiques |
Réponse à la colchicine, origine ethnique |
acidurie mévalonique en poussée |
Urticaire déclenché par le froid |
surdité |
Surdité, Méningite chronique à PMN, arthropathies |
Poussée de longue durée, éruption migrante |
|
FMF, fièvre méditerranéenne familiale ; HIDS, syndrome hypergammaglobulinemia D with periodic fewer syndrome, MWS, Muckle Wells syndrome ; FCAS , familial cold autoinflammatory syndrome ; CINCA, chronic infantile neurological cutaneous and articular syndrome ; TRAPS, tumor necrosis factor receptor-associated periodic syndrome syndrome ; PFAPA : periodic fever adenopathy pharyngitis aphtosis. AR :autosomique récessif ; AD : autosomique dominant ; +++ très fréquent, ++ fréquent, + occasionnel, +/- rare. |
|||||||
Références
Brydges S, Kastner D. The systemic autoinflammatory diseases: inborn errors of the innate immune system. Curr. Top. Microbiol. Immunol 2006,305:127-60.
Hull KM, Shoham N, Chae JJ et al. The expanding spectrum of systemic autoinflammatory disorders and their rheumatic manifestations. Curr Opin Rheumatol 2003;15: 61-9.
Samuels J, Ozen S. Familial Mediterranean fever and the other autoinflammatory syndromes: evaluation of the patient with recurrent fever. Curr Opin Rheumatol 2006;18:108-17.
Stojanov S, Kastner D. Familial autoinflammatory diseases: genetics, pathogenesis and treatment. Curr Opin Rheumatol 2005;17:586-99.