K. Fouatih, A. S. Lambert,
Service d’endocrinologie pédiatrique, CHU Bicêtre, AP-HP, Le Kremlin Bicêtre, France
Article validé par : Société Française d’Endocrinologie et Diabétologie Pédiatrique (SFEDP)
Introduction
Le retard pubertaire est un motif de consultation beaucoup plus fréquent chez le garçon (2,5% d’entre eux) que chez la fille.
Chez le garçon, il se caractérise par l'absence d'augmentation du volume testiculaire (inférieur à 4 mL ou avec une longueur testiculaire inférieure à 25 mm) après 14 ans (soit au-delà de +2 déviations standards par rapport à l’âge moyen du début de la puberté). Le développement de la pilosité pubienne, bien qu'indicatif de l'adrénarche, ne constitue pas en soi un signe du début de la puberté. Le rôle du pédiatre est de distinguer un retard pubertaire simple (le plus fréquent) d'un retard dû à une cause sous-jacente.
Chez la fille, il se caractérise par l’absence de développement des seins autour de 13 ans ou l’absence de règles (aménorrhée primaire) à 15 ans. Ici, il faut rechercher en priorité une cause organique. Un syndrome de Turner doit être évoqué en premier lieu devant tout retard pubertaire
associé à une petite taille. Le retard pubertaire simple est un diagnostic présomptif nécessitant une surveillance clinique.
Dans les deux sexes, l’arrêt de la progression de la puberté pendant plus de 3 ans doit faire évoquer une pathologie pubertaire.
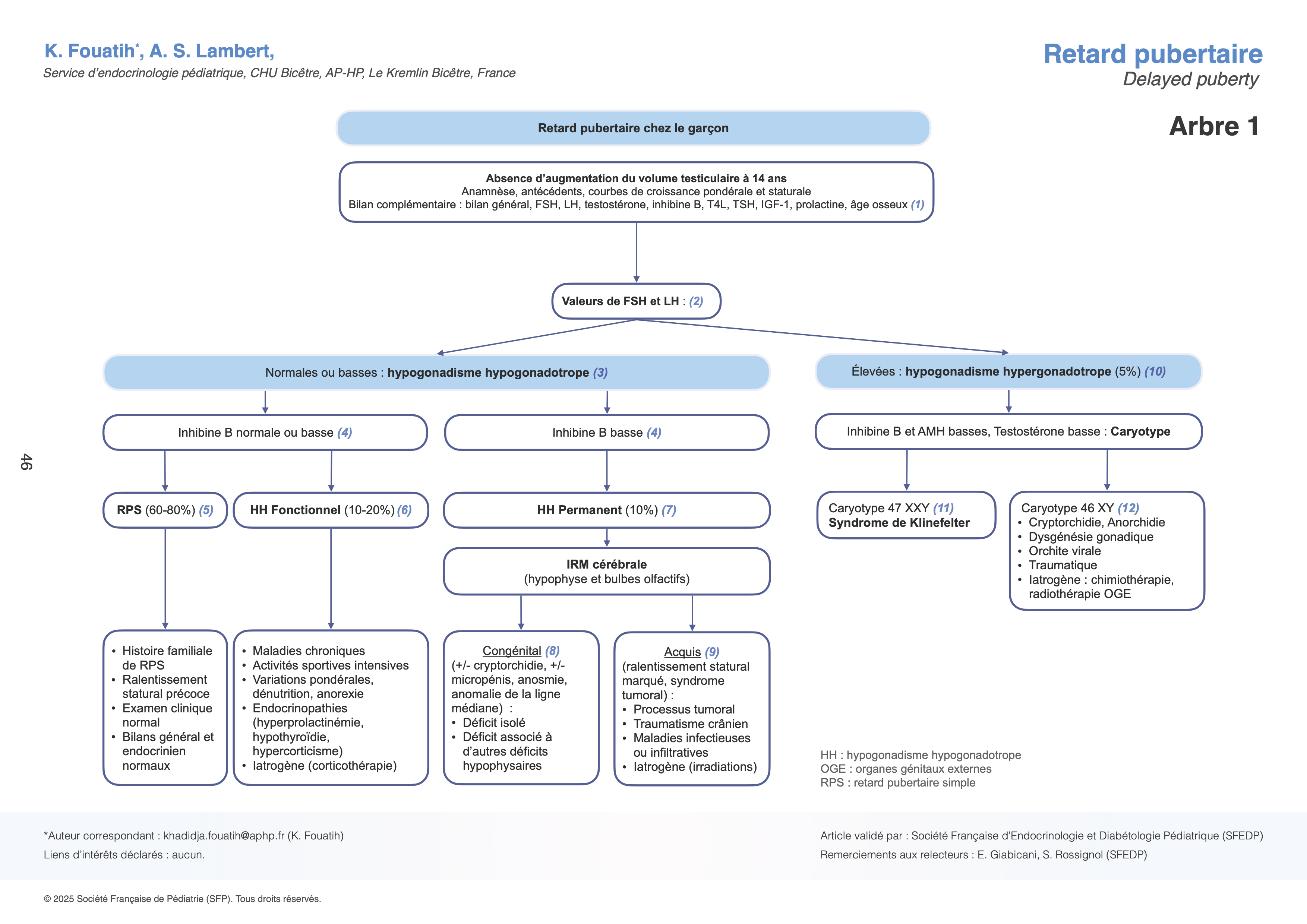
Conduite diagnostique devant un retard pubertaire chez le garçon (arbre 1)
(1) Les antécédents doivent être détaillés, notamment la taille et l’âge de la puberté chez les parents (faciles à repérer chez les femmes par l’âge des premières règles ou la notion de croissance tardive chez les apparentés masculins) et la fratrie. L’analyse de la courbe de croissance staturo-pondérale (à tracer) est indispensable. Les antécédents pathologiques et les traitements reçus (corticothérapie, chimiothérapie, radiothérapie, etc.) sont soigneusement notés. Sont recherchés à l’interrogatoire des troubles digestifs, une polyuro-polydipsie, des céphalées et une anomalie du champ visuel.
L’examen clinique doit rechercher les signes évoquant une pathologie acquise, une dysmorphie. L’examen des organes génitaux externes (cf. introduction) permettra de quantifier soit l’absence totale, soit un début passé inaperçu, ou encore l’arrêt de la puberté. Enfin, la présence d’une anosmie (incapacité à sentir les odeurs) permet d’évoquer, sur la seule clinique, le diagnostic rare d’hypogonadisme hypogonadotrope par syndrome de Kallmann de Morsier. Le bilan initial comporte un bilan de base à la recherche d’une pathologie générale et/ou inflammatoire (rénale, hépatique, systémique). La détermination de l’âge osseux permet d’obtenir un repère quantifiable de la maturation du squelette par les stéroïdes sexuels. Il est lu à l’aide d’un atlas radiologique (Greulich et Pyle), à partir d’une radiographie du poignet et de la main gauche. Le début de la puberté correspond habituellement à un âge osseux de 13 ans chez le garçon (apparition du sésamoïde du pouce).
(2) En première intention, les dosages plasmatiques des FSH et LH de base, couplés à ceux des stéroïdes sexuels, sont l’étape initiale essentielle du diagnostic étiologique.
(3) Des valeurs normales ou basses de gonadotrophines (FSH et LH) sont en faveur d’une origine hypothalamo-hypophysaire (hypogonadisme hypogonadotrope).
(4) Le taux d’inhibine B peut permettre d’orienter la suite du raisonnement.
(5) En présence de gonadotrophines basses, le diagnostic le plus courant (60 à 80% des cas ) est celui du retard pubertaire simple, qui correspond à un retard physiologique de l'activation de l'axe hypothalamo-hypophyso-gonadique (HPG). Il s'agit d'un diagnostic d'élimination. Le taux d'inhibine B est généralement supérieur 60 pg/mL. Lors de l'interrogatoire, on retrouve fréquemment un antécédent familial de retard pubertaire, ainsi qu'un ralentissement précoce de la vitesse de croissance, alors que les évaluations clinique et biologique sont toujours normales.
(6) L'hypogonadisme hypogonadotrope fonctionnel (HHF) représente jusqu'à 20% des cas et correspond à une inhibition temporaire de la maturation de l'axe HPG, causée par une maladie organique sous-jacente, une variation importante du poids, une activité physique intense et prolongée, ou encore une prise de médicaments tels que les corticoïdes. Toutefois, il existe une variabilité interindividuelle dans la réponse des jeunes à ces facteurs de stress, ce qui influence leur susceptibilité à développer un retard pubertaire. Dans ce cas, le déficit gonadotrope est réversible une fois que l'étiologie sous-jacente est corrigée.
(7) Un hypogonadisme hypogonadotrope permanent (HHP) est fortement suspecté devant une inhibine B basse (Se et Sp à 100% si < 35 pg/mL). Dans ce cas, des investigations supplémentaires, incluant une IRM cérébrale sont nécessaires pour rechercher une malformation congénitale ou une cause acquise.
(8) Une origine génétique est identifiée dans près de 50% des cas d’HHP congénital grâce aux panels de gènes (NGS) disponibles aujourd’hui. Le tableau clinique peut inclure une cryptorchidie bilatérale, un micropénis, une anosmie (syndrome de Kallmann de Morsier), ou encore des malformations rénales. Une résolution spontanée partielle est observée dans 10% des cas.
(9) L’HHP acquis est fréquemment associé à d'autres atteintes de l'antéhypophyse. Le diagnostic est guidé par les antécédents médicaux, chirurgicaux ou traumatiques, ainsi que par les signes cliniques tels qu’un syndrome tumoral, et par les anomalies détectées à l’IRM hypophysaire.
(10) Un taux élevé de gonadotrophines suggère un hypogonadisme hypergonadotrope, une situation représentant seulement 5% des cas de retard pubertaire. Dans ce cas-là, la testostérone ainsi que les marqueurs sertoliens (AMH, inhibine B) peuvent être normaux ou bas.
(11) Le caryotype constitue l’examen de première intention pour dépister le syndrome de Klinefelter (47XXY), qui en est la cause la plus fréquente. Cliniquement, il est souvent retrouvé à cet âge une grande taille, une gynécomastie et un faible volume testiculaire. Un retard de langage et des difficultés d’apprentissage sont parfois présents.
(12) D’autres causes d’hypogonadisme hypergonadotrope à caryotype 46, XY incluent la cryptorchidie, la dysgénésie gonadique, ainsi que des facteurs traumatiques, infectieux ou iatrogènes subis par les organes génitaux externes.