W. Abou Chahla¹, C. Leblanc²
¹Service d’hématologie pédiatrique, hôpital Jeanne de Flandre, CHU Lille, CEREO, Lille, France
²Service de pédiatrie générale, hôpital mère enfant, CHU de Nantes, Nantes, France
Article validé par : Groupe de Pathologie Tropicale (GPTrop), Société d’Hématologie et Immunologie Pédiatrique (SHIP), Société Pédiatrique de Pneumologie et Allergologie (SP2A).
Introduction
L’hyperéosinophilie (HE) est définie par un taux d’éosinophiles sériques circulants > à 500/mm³. Il s’agit d’une anomalie biologique fréquente en pédiatrie, pouvant être bénigne et transitoire ou révéler des pathologies sous-jacentes plus sévères.
L’HE peut être classée en trois catégories en fonction de son importance :
• Faible (500–1500/mm³) : souvent bénigne et réversible.
• Modérée (1500–5000/mm³) : nécessitant une recherche étiologique plus approfondie.
• Sévère (> 5000/mm³ ou 5 G/L) : associée à un risque élevé d’atteinte viscérale (Syndrome Hyperéosinophilique, SHE), nécessitant une prise en charge urgente.
Plutôt que d’aborder l’HE uniquement par son étiologie (hyperéosinophilie primitive, secondaire et idiopathique), ce document privilégie une approche basée sur les conséquences cliniques. L’objectif est d’identifier précocement les formes graves, notamment le SHE, afin d’initier une prise en charge rapide et adaptée.
Le point commun entre éosinophiles et augmentation des IgE est l’activation de la réponse immunitaire Th2. L’IL-5 est produite par les lymphocytes Th2 qui vont permettre la production, la maturation, l’activation et la survie des éosinophiles. D'autres cytokines interviennent comme IL-4 et IL-13, qui stimulent la commutation isotypique des lymphocytes B vers la production d’IgE. On peut retenir trois causes principales d’augmentation des éosinophiles et des IgE : les maladies atopiques (asthme, eczéma, rhinite allergique…), les parasitoses helminthiques, certaines maladies auto-inflammatoires ou des syndromes rares (SHE lymphoïdes).
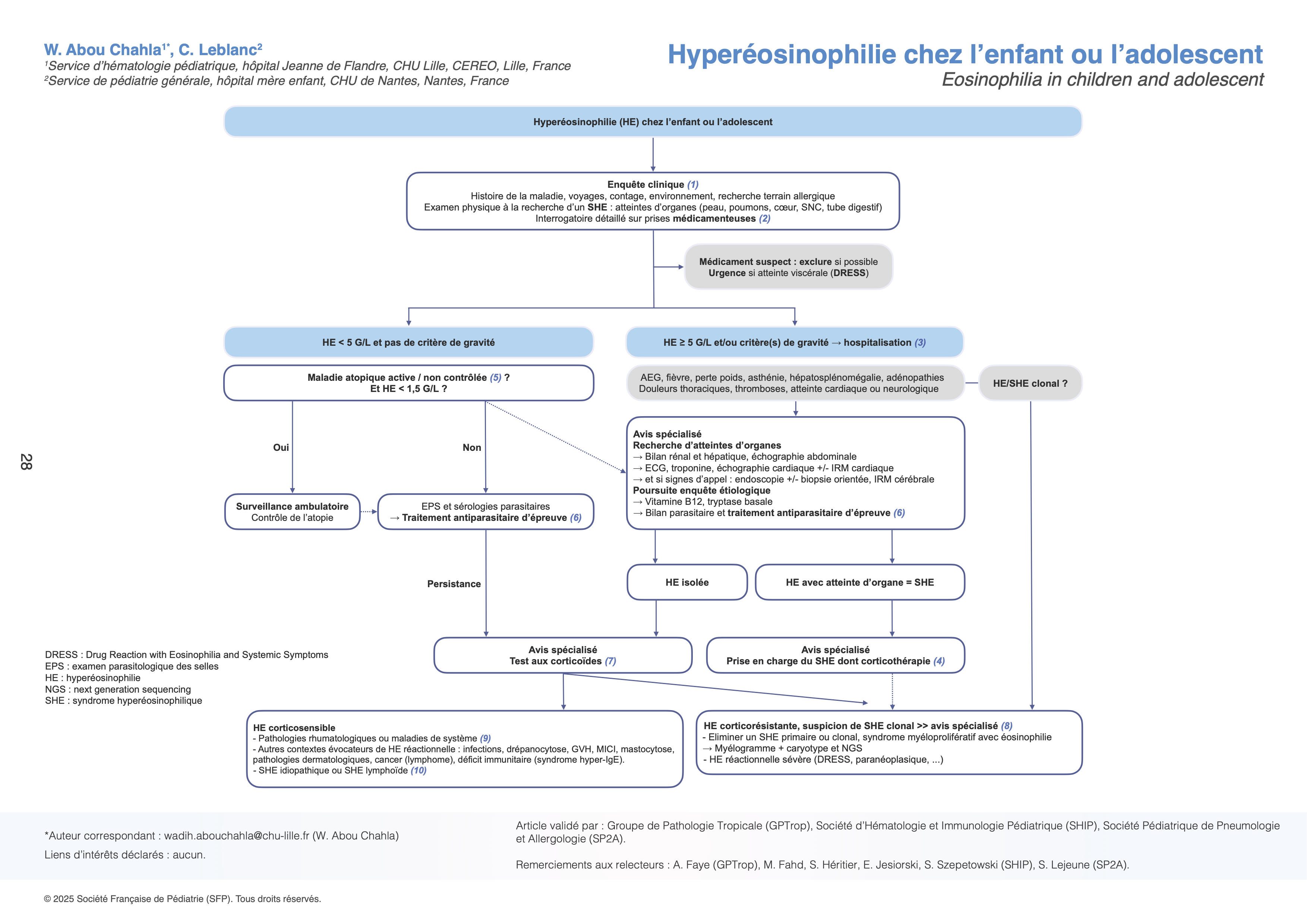
Démarche diagnostique face à une hyperéosinophilie
(1) Enquête clinique
À l’interrogatoire, préciser :
• Si possible : ancienneté de l’HE et évolution des taux d’éosinophiles.
• Symptômes associés : prurit, éruption cutanée, angiœdème, signes digestifs, fatigue, fièvre, sueurs nocturnes, amaigrissement, douleurs musculaires ou articulaires.
• Voyages récents ou exposition à des agents infectieux : antécédents de voyage en zone d’endémie parasitaire, contact avec des animaux, alimentation (consommation de viande crue, poissons crus, eau non traitée, produits de la cueillette / du potager hors industrie agro-alimentaire contrôlée), prurit anal et perturbation du sommeil chez les jeunes enfants.
• Terrain atopique : asthme, rhinite allergique, dermatite atopique, allergies alimentaires.
• Prise médicamenteuse récente : notamment antibiotiques, antiépileptiques, AINS ou autres médicaments pouvant induire une HE.
À l’examen clinique, rechercher :
• Une atteinte cutanée ou muqueuse (lésions prurigineuses, dermatite atopique, urticaire, purpura).
• Une atteinte pulmonaire (dyspnée, sibilants).
• Une atteinte cardiaque (souffle, signes d’insuffisance cardiaque).
• Une atteinte neurologique (signes d’encéphalopathie, neuropathie périphérique).
• Un syndrome tumoral (hépatosplénomégalie, adénopathies).
(2) Exclure une cause médicamenteuse
Formes cliniques principales :
Hyperéosinophilie médicamenteuse isolée
Des médicaments peuvent provoquer une HE modérée et transitoire sans atteinte systémique. L’arrêt du médicament entraîne généralement une normalisation rapide de l’éosinophilie.
Syndrome DRESS
Réaction d’hypersensibilité retardée, potentiellement grave, associant :
• Tableau systémique : fièvre prolongée, altération de l’état général et atteinte cutanée (80 à 90% des cas) : exanthème maculopapuleux étendu, parfois prurigineux, œdème du visage et des extrémités, pustulose stérile, purpura, décollement épidermique.
• Atteinte hépatique (50 à 80% des cas) : cytolyse hépatique, hépatite fulminante exceptionnelle mais possible.
• Atteintes d’organe possibles : néphrite interstitielle, pneumopathie éosinophilique, myocardite.
• Hyperéosinophilie sanguine apparaissant parfois secondairement.
Délai de survenue variable, classiquement de 2 à 8 semaines après le début d’exposition (mais délais plus courts décrits).
Principales classes thérapeutiques impliquées :
• Allopurinol.
• Anticonvulsivants : carbamazépine, phénytoïne, lamotrigine, phénobarbital.
• Antibiotiques : bêta-lactamines, sulfamides, vancomycine, rifampicine.
• AINS et antalgiques : ibuprofène, naproxène, paracétamol.
• Antituberculeux : isoniazide, éthambutol.
Prise en charge :
• Arrêt immédiat du médicament suspect ; la normalisation de l’HE peut prendre plusieurs semaines.
• En cas d’atteinte viscérale sévère (DRESS, atteinte hépatique, myocardite, pneumopathie éosinophilique sévère) :
o Corticothérapie systémique (0,5-1 mg/kg/j de prednisone, avec décroissance progressive).
o Suivi rapproché des fonctions hépatiques, cardiaques et pulmonaires.
o Avis allergologique, contre-indication du médicament impliqué.
Hyperéosinophilie ≥ 5 G/L et/ou critères de gravité → hospitalisation
(3) Des critères de gravité doivent-être recherchés :
• Signes d’atteinte viscérale : douleur thoracique, dyspnée, signes d’insuffisance cardiaque évoquant une myocardite, fibrose cardiaque, thrombose, atteinte neurologique.
• Signes systémiques : altération de l’état général avec fièvre et amaigrissement.
• Hépatosplénomégalie, adénopathies ou signes sur la NFS en faveur d’une hémopathie (anémie, thrombopénie, macrocytose, blastose).
Une hospitalisation est alors requise, permettant d’effectuer des investigations approfondies et d’initier rapidement la prise en charge adaptée. Un avis spécialisé est nécessaire.
En cas de signe évocateur d’une hémopathie (altération de l’état général, hépatosplénomégalie, cytopénie, myélémie), aller directement à l’étape (8) avec la réalisation d’un myélogramme.
Quelle que soit sa cause, l’HE peut s’accompagner d’une atteinte d’un ou plusieurs organes, on parle alors de Syndrome Hyperéosinophilique (SHE) :
• Thrombose : événements thrombotiques artériels ou veineux.
• Atteinte cutanée et muqueuse : prurit, urticaire, dermatite atopique, prurigo, angiœdème, ulcérations, purpura.
• Atteinte neurologique : atteinte centrale (encéphalopathie, troubles cognitifs) ou périphérique (neuropathies).
• Atteinte digestive ou hépatique : diarrhées, douleurs abdominales, dysphagie, ascite à éosinophile, ictère.
• Atteinte respiratoire : atteinte bronchique ou pulmonaire.
• Atteinte cardiaque : douleur thoracique, insuffisance cardiaque droite ou gauche.
Le bilan à la recherche d’atteintes d’organes repose sur un interrogatoire précis, un examen clinique complet (notamment cardiopulmonaire, cutané et neurologique) et des explorations complémentaires ciblées : bilan rénal et hépatique ; échographie abdominale (splénomégalie ?), ECG, troponine, échographie cardiaque +/- IRM cardiaque ; endoscopie +/- biopsie orientée si atteinte viscérale ; IRM cérébrale sur signe d’appel. ! ⚠️ L’atteinte cardiaque doit faire l’objet d’un dépistage systématique, car elle peut être asymptomatique aux premiers stades et conduire à des complications emboliques et une fibrose myocardique irréversible si elle n’est pas diagnostiquée précocement.
L’enquête étiologique doit être poursuivie en parallèle : dosage de vitamine B12, tryptase sérique, bilan parasitaire et traitement antiparasitaire d’épreuve (voir (5)).
(4) La prise en charge d’un SHE avec atteinte d’organe repose sur le traitement des atteintes et une corticothérapie 1 à 2 mg/kg/j avec pour objectif la rémission des manifestations cliniques et biologiques.
HE < 5 G/L et aucun critère de gravité : prise en charge ambulatoire, confirmationet classification de l’HE
La recherche d’une cause sous-jacente est nécessaire, même en l’absence de symptômes évidents (infection parasitaire, allergies, terrain atopique, asthme…).
(5) Rechercher des manifestations cliniques orientant vers une origine atopique.
L’atopie et ses manifestations reliées peuvent être associées à une HE habituellement faible (500-1500/mm3) : dermatite atopique, asthme, rhinite allergique, allergies alimentaires, urticaire chronique, certaines atteintes gastro-intestinales dont l’œsophagite à éosinophiles. Ces signes sont habituellement connus et/ou aisément identifiables, au besoin après évaluation avec un allergologue. Il ne sera pas nécessaire de poursuivre les investigations à ce stade si HE < 1,5 G/L. Un contrôle de la maladie atopique s’accompagnera d’une baisse des éosinophiles.
En l’absence d’atopie :
• HE ≤ 1,5 G/L : surveiller en ambulatoire, contrôler la NFS à 1 mois ; en cas de persistance, recherche une cause infectieuse (parasitaire).
• HE > 1,5 G/L : rechercher une atteinte d’organes (notamment cardiaque car certaines atteintes peuvent être asymptomatiques au début) et poursuivre l’enquête étiologique.
(6) Rechercher une cause infectieuse (parasitaire avant tout)
L’HE peut être secondaire à une infection parasitaire, notamment en cas de voyage en zone d’endémie ou d’exposition à des vecteurs parasitaires dans un contexte domestique.
La prévalence de l’étiologie parasitaire en cas d’hyperéosinophilie est différente si contexte de séjour en zone d’endémie (majorité des causes) ou non (1-10%). En pratique, l’efficacité du traitement d’épreuve antiparasitaire bien conduit permet de diminuer le taux d’éosinophiles de moitié dans les semaines qui suivent ce traitement dans 30 à 60% des cas.
Une anamnèse détaillée est essentielle pour orienter les investigations et ne pas passer à côté d’une parasitose évolutive :
• Habitudes alimentaires : consommation de viande crue ou mal cuite (porc, bœuf, gibier, poisson), crudités mal lavées, eau non traitée.
• Loisirs et activités : baignades en eau douce tropicale (risque de schistosomose), randonnées pieds nus(ankylostomiase), chasse (trichinellose).
• Contacts avec des animaux : chiens et chats (toxocarose), rongeurs (angiostrongylose), poissons et crustacés crus (anisakiase).
• Voyages en zone d’endémie parasitaire : pays tropicaux, Asie, Afrique, Amérique du Sud.
Les phases de primo-invasion des helminthiases sont souvent associées à une hyperéosinophilie transitoire, avec des taux élevés d’IgE totales en raison de la migration tissulaire du parasite. Les sérologies et les examens parasitologiques des selles (EPS) ou des urines (EPU) doivent être répétés à distance si la suspicion clinique est forte, car ils peuvent être négatifs au début de l’infestation. Les protozooses (paludisme, leishmaniose, amibiase) ne provoquent pas d’hyperéosinophilie et doivent être évoquées sur d’autres arguments cliniques.
Les oxyuroses peuvent être responsables d’hyperéosinophilie faible. Elles sont cosmopolites et fréquentes chez les jeunes enfants. Un traitement peut être proposé en cas de point d’appel clinique (prurit anal, irritation vulvaire chez la fille, troubles du sommeil). Après prise en charge, les symptômes et l’HE doivent s’amender rapidement. Les oxyuroses ne sont pas responsables d’HE modérée et/ou sévère ; situations devant conduire à la recherche d’un autre diagnostic.
En l’absence de voyage en zone d’endémie (parasites cosmopolites)
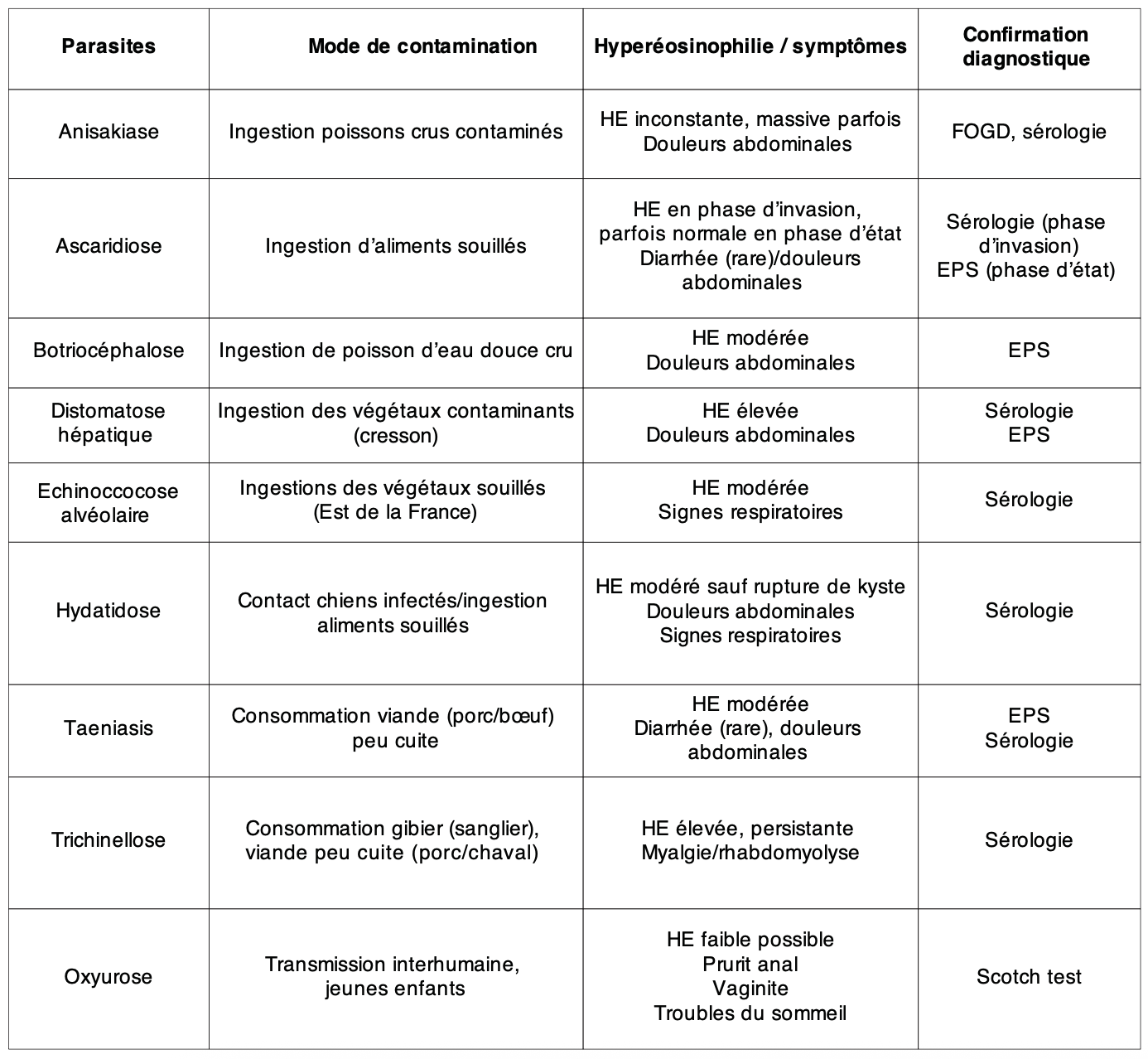
Examens de première intention :
• EPS x 3 (sur 10 jours) : recherche d’helminthes en phase d’état (2-3 mois après la primo-invasion).
• Scotch test : recherche d’oxyurose.
• Sérologie toxocarose (exposition aux animaux).
• Autres sérologies à discuter selon le contexte.
Traitement antiparasitaire d’épreuve (sans voyage en zone d’endémie)
• HE < 1,5 G/L sans cause évidente → traitement empirique par :
- Flubendazole : 100 mg/j pendant 3 jours, avec rappel d’une monodose, 15 jours plus tard.
- Albendazole : 200 mg (< 2 ans) ou 400 mg/j pendant 1-3 jours, avec rappel d’une monodose à J15.
- Cibles principales : oxyurose, ascaridiose.
• Émission d’anneaux parasitaires spontanés dans les selles ou par l’anus → traitement par :
- Praziquantel : 15 mg/kg en une prise unique.
- Cibles principales : ténia, botriocéphalose, hyménolépiase.
• HE > 1,5 G/L inexpliquée → traitement empirique par :
- Albendazole : 10-15 mg/kg/j (max 800 mg/j) en 2 prises pendant 10-15 jours.
- Cibles principales : toxocarose, trichinellose, ascaridiose (en cas de sérologie toxocarose positive : éliminer une atteinte oculaire parasitaire avant traitement).
En cas de voyage en zone d’endémie
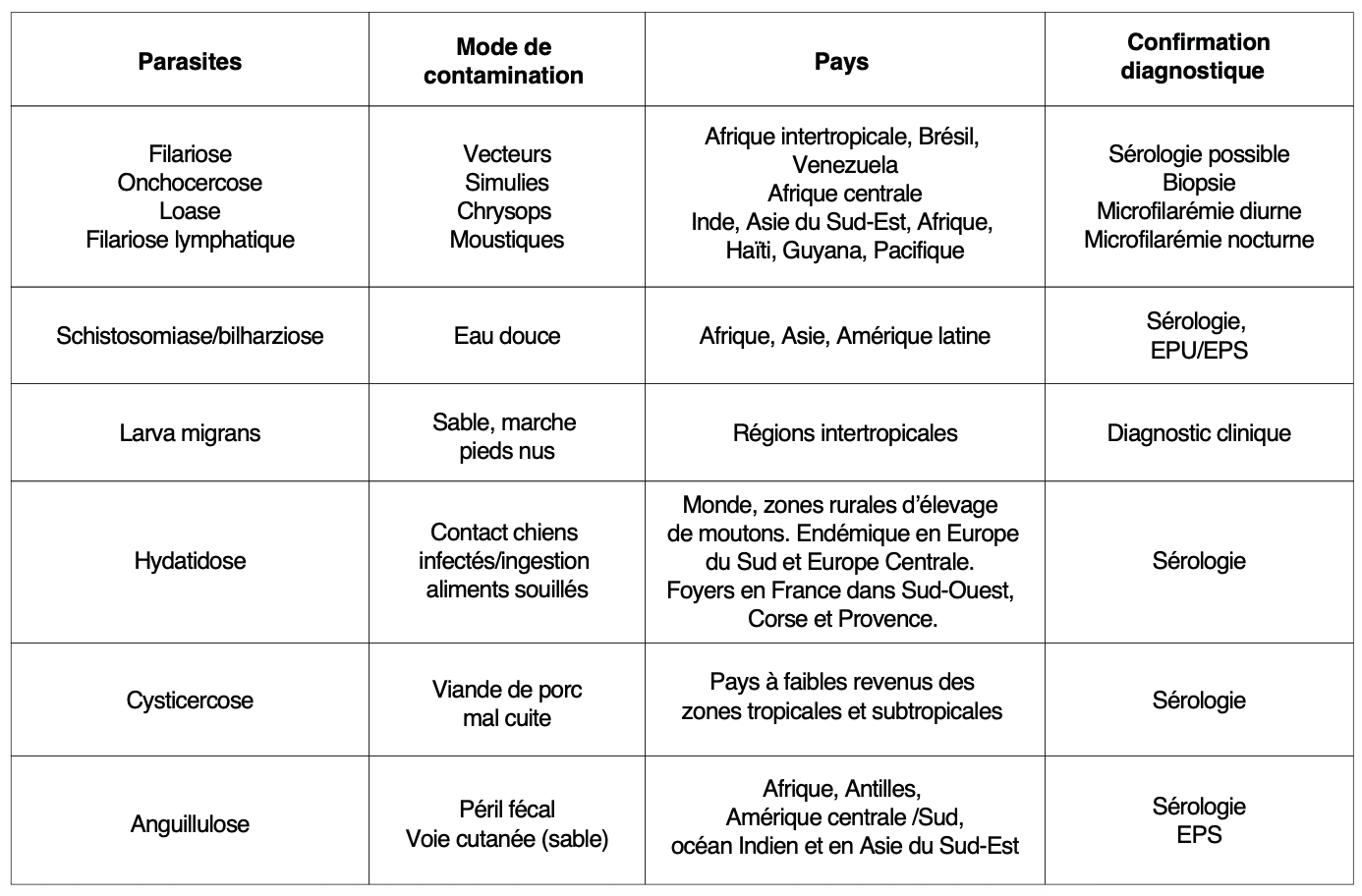
Avis spécialisé pour adapter la conduite diagnostique et thérapeutique.
Infection possible à partir d’un retour de voyage datant d’au moins 3 mois et pouvant aller jusqu’à plusieurs années.
Traitement antiparasitaire d’épreuve après voyage en zone d’endémie uniquement après enquête et discussion avec un parasitologue :
• J1 : Ivermectine 200 µg/kg à jeun (éventuellement une seconde prise à J2 ou J15 si confirmation d’anguillulose).
o Cibles principales : anguillulose (présente aussi en Europe), filariose.
• J2 : Praziquantel 40 mg/kg en une prise après le repas du soir.
o Cibles principales : bilharzioses, distomatoses.
• J3 à J18 : Albendazole 10-15 mg/kg/j (max 800 mg/j) en 2 prises pendant 3-15 jours.
o Cibles principales : toxocarose, ankylostomiase, ascaridiose, trichocéphalose.
Un suivi à distance de l’hyperéosinophilie est recommandé. Il est fréquent d’observer une HE se majorant transitoirement quelques jours à semaines après le traitement des helminthiases ; elle doit régresser dans les 3 mois après le début du traitement.
Autres causes infectieuses non parasitaires :
• Certaines infections virales transitoires peuvent être responsables d’une HE (EBV, CMV, HHV6), mais elles se résolvent généralement spontanément.
• L’infection par le VIH doit être systématiquement recherchée si HE persistante.
• L’infection par HTLV1 doit être évoquée chez les patients originaires ou ayant séjourné en zone endémique (Japon, Caraïbes, Afrique sub-saharienne).
• La tuberculose peut être associée à une HE, nécessitant un test de détection de l'interferon gamma ou une IDR en cas d’adénopathies associées.
• Une aspergillose bronchopulmonaire allergique doit être recherchée en cas d’atteinte respiratoire (IgE totales > 500 KUI/L, bronchiectasies proximales au scanner thoracique, dosage d’IgG et IgE anti-aspergillaires).
(7) Test thérapeutique aux corticoïdes
Dans le bilan d’une HE inexpliquée et persistante après un traitement antiparasitaire d’épreuve, un avis spécialisé est nécessaire. Un test thérapeutique aux corticoïdes évalue la normalisation du nombre de PNE après 3 jours de corticothérapie à 0.5 mg/kg/j d’équivalent prednisone. Il permet d’éliminer une HE ou un SHE primaire et de s’assurer de l’efficacité des corticoïdes pour une poussée ultérieure.
(8) HE ou SHE clonale ou primaire, à suspecter en cas de corticorésistance
Les hyperéosinophilies d’origine clonale sont rares, prédominantes chez l’homme, et exceptionnelles chez l’enfant, avec seulement quelques cas cliniques rapportés dans la littérature. En France, leur incidence est estimée à moins de 1 cas par an.
À évoquer devant :
• Signes cliniques : altération de l’état général, hépatosplénomégalie, adénopathies.
• Anomalies hématologiques : cytopénies ou, à l’inverse, thrombocytose ou polyglobulie ; monocytose ou basophilie associée ; myélémie.
• Elévation de la vitamine B12, augmentation de la tryptase sérique.
• Corticorésistance.
Il est nécessaire de référer à une équipe spécialisée qui évaluera l’éventualité diagnostique et proposera une recherche sur prélèvement sanguin des transcrits FIP1L1-PDGRFα, BCR-ABL, et mutation JAK2 V617F, médullogramme et étude du caryotype médullaire +/- NGS.
Autres causes rares d’HE
(9) Causes rhumatologiques, maladies de système
L’hyperéosinophilie peut être secondaire à certaines pathologies rhumatologiques, bien que l’éosinophilie ne soit pas toujours au premier plan. Par exemple, en cas de manifestation articulaire, il faudra rechercher une polyarthrite rhumatoïde ou une arthrite juvénile idiopathique. En cas de polypose nasale et d’asthme, il faudra rechercher une granulomatose éosinophilique avec polyangéïte.
D’autres pathologies s’accompagnent de HE réactionnelle : infections (voir fin du paragraphe (6)), drépanocytose, GVH, MICI, mastocytose, pathologies dermatologiques rares (cellulite à éosinophiles paniculite, pustulose,…), cancer (lymphome), déficit immunitaire (Syndrome hyperIgE).
(10) Syndrome Hyperéosinophilique (SHE) dit "lymphoïde" :
Le SHE lymphoïde est une cause rare d’hyperéosinophilie, résultant d’une expansion clonale de lymphocytes T anormaux, qui produisent des cytokines favorisant une différenciation et une activation éosinophilique accrue, notamment via l’IL-5. Chez l’enfant, ce syndrome est exceptionnel et ne doit pas être recherché en première intention. Toutefois, il peut être envisagé en cas d’hyperéosinophilie persistante inexpliquée, surtout si elle est associée à des signes évocateurs : HE isolée, signes allergiques, adénomégalies, hépatosplénomégalie. Ces SHE sont cortico-sensibles et un phénotypage lymphocytaire dans un laboratoire spécialisé guidera la démarche diagnostique.
Conclusion
L’hyperéosinophilie (HE) est un syndrome biologique hétérogène pouvant être associé à une grande variété de pathologies, allant de causes bénignes et transitoires à des maladies graves nécessitant une prise en charge spécifique.
L’objectif de ce guide est d’aider à structurer l’analyse clinique et biologique afin de ne pas méconnaître une HE grave, tout en évitant des investigations inutiles dans les formes bénignes. La reconnaissance précoce des formes sévères et potentiellement dangereuses est essentielle pour éviter des complications systémiques irréversibles.
Les étiologies infectieuses, allergiques, immunologiques, hématologiques et syndromiques doivent être envisagées de manière méthodique, en adaptant le bilan à la présentation clinique du patient. La distinction entre hyperéosinophilie primitive, secondaire et idiopathique (HE persistantes inexpliquées) est un enjeu central du diagnostic, nécessitant parfois des explorations approfondies, incluant des examens médullaires ou immunologiques.
Face à une hyperéosinophilie persistante ou sévère, une approche rigoureuse et multidisciplinaire est indispensable, impliquant parfois des spécialistes en hématologie, immunologie, allergologie ou infectiologie.