
Vous êtes ici
Thématique:
Conduite diagnostique devant une dysmorphie
A. Goldenberg
Service de génétique médicale, hôpital Charles-Nicolle, 1, rue de Germont, 76031 Rouen cedex, FranceCorrespondance - Adresse e-mail : alice.goldenberg@chu-rouen.fr (A. Goldenberg).
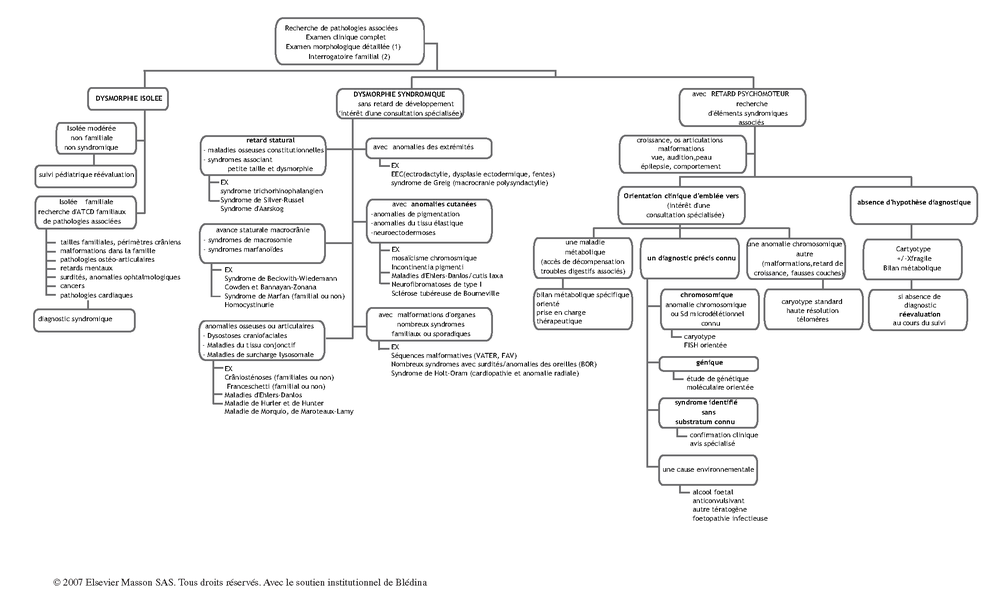
Arbre décisionnel – Commentaires
(1) L’examen morphologique doit être complet (crâne, face, extrémités, thorax, abdomen, organes génitaux, ostéoarticulaire, examen endobuccal, peau et phanères). Il obéit à des critères précis mais doit tenir compte des variations individuelles et des anomalies mineures retrouvées dans la population générale :
-
examen du crâne : forme du crâne, palpation des sutures, taille des fontanelles (craniosténoses, défaut d'ossification), zones d'alopécie, implantation des cheveux (syndrome de Turner ou de Noonan) ;
-
examen de la face : rigoureux et systématique, détaille la forme générale du visage puis chaque élément de haut en bas ;
-
région périorbitaire : distance inter-orbitaire (hypertélorisme dans la maladie du cri-du-chat, hypo-télorisme dans les holoprosencéphalies), forme des sourcils (marqués fournis et jointifs (synophris) dans le syndrome de Cornélia de Lange), largeur et orientation des fentes palpébrales (étroites dans la microdélétion 22q11) ;
-
étage moyen de la face :
-
étude du nez : racine (déprimée en ensellure dans l’achondroplasie, saillante en casque grec dans la microdélétion 4p), os propres, arête nasale (aspect en bec dans le syndrome de Seckel), pointe (bulbeuse dans le syndrome trichorhinophalangien), éversion des narines et la forme de la columelle (saillante dans le syndrome Rubinstein-Taybi) ;
-
le relief malaire doit être palpé (hypoplasie malaire du syndrome de Franceschetti) ;
-
étude de la forme et du relief du philtrum (effacé dans le syndrome d’alcoolisation fœtale) ;
-
examen de la bouche : fentes labiales, épaisseur des lèvres (charnues dans syndrome de Williams et Beuren), taille de la bouche et sa forme. En endobuccal : palais (fente, palais ogival), luette, gencives (hypertrophie gingivale dans les maladies de surcharge lysosomale, freins gingivaux dans les syndromes oro-facio-digitaux), langue (macroglossie, glossoptose, langue lobulée), dents (nombre et morphologie).
-
-
étage inférieur de la face : forme et taille du menton (micrognatisme), position (rétrognatisme, prognatisme).
-
examen des oreilles : position (implantation normale si la ligne horizontale passant par le canthus interne de l'œil et l'occiput coupe l'oreille au niveau de son tiers supérieur), orientation (le grand axe de l'oreille doit faire un angle de moins de 20° avec la verticale), taille (microtie), forme (ronde, triangulaire, présence ou non d'un lobule), ourlage. Éléments associés : condylomes prétragiens (syndrome de Goldenhar), fistules pré-auriculaires (syndrome branchio-oto-rénal), encoches sur ou derrière les lobes (Wiedemann-Beckwith).
-
examen des extrémités : anomalies de nombre des doigts (polydactylie, oligodactylie, ectrodactylie), fusions des doigts (syndactylies osseuses ou membraneuses totales ou mineures), limitations de mobilité (camptodactylie, arthrogrypose), anomalies du pouce (pouce digitalisé, triphalangé, d'implantation proximale), pouce bifide ou large dans le syndrome de Rubinstein-Taybi, pouce hypoplasique dans l'anémie de Fanconi. Longueur des doigts (brachydactylie, arachnodactylie), et des métacarpiens. Plis palmaires (unique transverse ou équivalent), plis de flexion interphalangiens (absence d'un pli, plis non parallèles avec inclinaison du doigt dans les clinodactylies). Le type d'anomalie des doigts peut orienter vers l'identification du mécanisme (aspects d'amputation précoce disruptif dans les cas de brides amniotiques). Les anomalies des doigts peuvent être évolutives (mains en griffe dans les maladies de surcharge lysosomales, macrodactylies dans le syndrome de Protée, acro-ostéolyses dans la progeria).
-
examen cutané : anomalies de pigmentation (mosaïcisme chromosomique, incontinentia pigmenti), taches café-au-lait et neurofibromes (neurofibromatose type I), vergetures (Marfan), cutis laxa, angiofibromes et fibromes périunguéaux (Bourneville).
(2) Enquête familiale : arbre généalogique sur deux à trois générations. Recherche de consanguinité. Chez les apparentés : antécédents malformatifs, fausses couches, enfants décédés, handicaps psychomoteurs, autres pathologies (sensorielles, neurologiques, cardiaques, anomalies de croissance, troubles orthopédiques, cancers…). Elle permet d’établir le mode de transmission de la pathologie et le don des indices diagnostiques chez le cas index.
Références
Goldenberg A, Saugier-Veber P. Retards mentaux d’origine génétique. Encyclopédie médico-chirurgicale : pédopsychiatrie [37-219-C-80].
Jones KL. Smith’s recognizable pattern of human malformation. Ed WB Saunders, 5th ed, 1997.
Orphanet : www.orpha.net