
Vous êtes ici
Thématique:
Purpura de l’enfant
![]()
W. Abou Chahla1*, E. Jeziorski2, F. Dubos3
1Service d’hématologie pédiatrique, CS 70001, CHU Lille, Hôpital Jeanne-de-Flandre, F-59000 Lille, France2Département urgence posturgences, Unité INSERM U 1058, université de Montpellier, CHU Montpellier, France
3Hôpital Roger-Salengro, urgences pédiatriques et maladies infectieuses et EA2694, F-59 000 Lille, CHU Lille, université Lille, France *Auteur correspondant - Adresse e-mail : wadih.abouchahla@chru-lille.fr (W. Abou Chahla).
| Article validé par : | |
|
SFP (Société Française de Pédiatrie) GFRUP (Groupe Francophone de Réanimation et Urgences Pédiatriques) SHIP (Société d’Hématologie et d’Immunologie Pédiatrique) |
|
Introduction
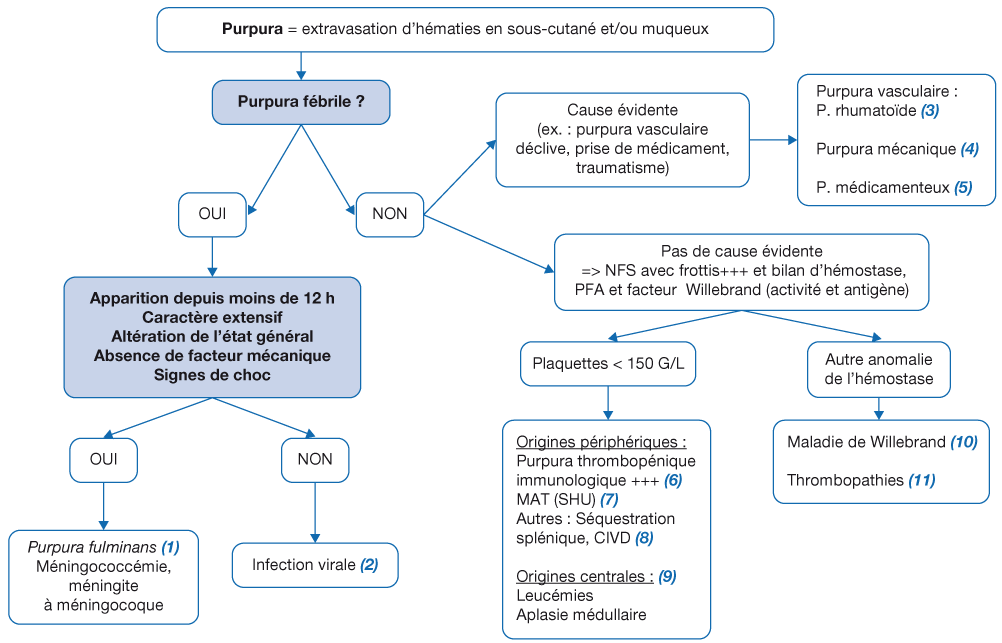
Le purpura est une extravasation de sang dans le derme ou le tissu muqueux. Il peut être lié à un simple traumatisme, mais peut aussi être le signe d’une pathologie sévère mettant en jeu le pronostic vital. Une évaluation minutieuse et précoce du patient permettra d’orienter la démarche diagnostique. Elle se base sur l’observation des lésions cutanéo-muqueuses (pétéchies, ecchymoses, nécroses, bulles hémorragiques), le nombre de lésions (<= ou > 100 pétéchies, ou <= ou > 5 ecchymoses), leux localisation (déclive, muqueuse), leur palpation (caractère maculeux, infiltré), leur caractère rapidement extensif, ainsi que la recherche de signes de choc septique et d’un syndrome tumoral. L’interrogatoire à la recherche d’antécédents personnels ou familiaux, et de signes hémorragiques complétera l’évaluation initiale.
La physiopathologie distingue deux cadres nosologiques : la rupture de l’intégrité vasculaire d’une part (infection, vascularite, traumatisme), et une anomalie de l’hémostase d’autre part (thrombopénie, anomalie des fonctions plaquettaires). Les examens complémentaires et les traitements dépendront de l’orientation diagnostique.
Orientation diagnostique devant un purpura de l’enfant
Le premier élément d’orientation est la notion de fièvre, et plus largement la présence de signes associés orientant vers une origine infectieuse. Si c’est le cas, deux diagnostics s’opposent.
(1) Purpura fulminans : infection invasive à méningocoque ou à pneumocoque, essentiellement. C’est le premier diagnostic à évoquer en cas de purpura fébrile. Il doit être considéré en cas de troubles hémodynamiques ou fièvre mal tolérée. Il doit être évoqué comme diagnostic par défaut en cas de purpura et de fièvre datant de moins de 12 h, et cela même si l’examen clinique et la biologie initiale sont rassurants à ce stade. Il faut identifier les signes de choc : tachycardie, temps de recoloration cutanée (TRC) >= 3 secondes, marbrures, pouls filant, trouble de conscience, cyanose, oligurie. Le caractère extensif est également un signe d’appel. C’est une urgence diagnostique et thérapeutique, nécessitant l’administration immédiate d’une Céphalosporine de 3e génération (C3G) IM/IV (Céfotaxime 50 mg/kg), un remplissage vasculaire par des solutés balancés, une prise en charge selon l’algorithme du choc septique et un transfert médicalisé en unité de réanimation.
En cas de purpura fulminans confirmé, un déficit en complément, en properdine ou une asplénie seront à rechercher au décours de la prise en charge urgente. En cas d’infection à un autre pathogène que le méningocoque (streptocoque du groupe B, pneumo coque, staphylocoque doré, varicelle, etc.), il faudra en plus rechercher un déficit en protéine S ou protéine C. À noter, si les hémocultures restent négatives au cours de la prise en charge initiale, les biopsies de lésions de purpura ecchymotiques ou nécrotiques ont une très bonne sensibilité pour la recherche du méningocoque.
Remarque : la méningite à méningocoque et la méningo coccémie sont également des urgences diagnostiques et thérapeutiques. Elles sont souvent associées à un purpura pétéchial ou ecchymotique. Les signes méningés et/ou de fièvre mal tolérée sont donc également à rechercher, et l’hémoculture est indispensable au moindre doute. Une antibiothérapie dans l’heure qui suit le début de la prise en charge est recommandée.
(2) À l’opposé, la grande majorité des purpuras fébriles sont bénins et liés le plus souvent à un épisode viral simple. Certains éléments cliniques peuvent être considérés comme rassurants, comme l’aspect pétéchial stable dans le temps, ou le caractère mécanique du purpura (topographie au niveau du visage et du haut du thorax dans un contexte de toux et/ou de vomissements). Les patients doivent être surveillés dans les services d’urgences ou en unité d’hospitalisation de courte durée (UHCD), avec un bilan (numération de la formule sanguine [NFS], C-reactive protein [CRP], procalcitonine, taux de prothrombine-temps de céphaline activée [TP-TCA]) quasi systématique et qui doit parfois être contrôlé pour écarter les rares cas de bactériémie et de méningite à méningocoque ou à pneumocoque, selon la précocité de réalisation du bilan initial par rapport au début des symptômes. Le bilan de coagulation, si pratiqué, peut mettre en évidence un allongement isolé du TCA lié à la présence d’un anticoagulant circulant.
En l’absence de contexte infectieux, l’interrogatoire et l’examen clinique permettent parfois d’orienter vers certaines étiologies, et sont le plus souvent complétés par un bilan sanguin.
(3) Purpura rhumatoïde : le diagnostic est avant tout clinique et repose sur la triade purpura (symétrique, vasculaire et déclive), manifestations articulaires (symétriques, inconstantes) et douleurs abdominales (inconstantes). L’atteinte rénale (pronostique) doit être recherchée par une bandelette urinaire avec une surveillance régulière pendant 1 an.
(4) Purpura mécanique : fréquent chez l’enfant, notamment en péri-orbitaire (effort de vomissement ou de toux), ou sur un membre par compression (garrot). Une atteinte sous- diaphragmatique ou déclive doit faire réviser ce diagnostic.
(5) Origine médicamenteuse : prise d’antiagrégant plaquettaire, de types anti-inflammatoires non stéroïdiens ou aspirine.
Lorsque l’orientation clinique n’est pas évidente, les pathologies qui suivent sont à rechercher avec un bilan de 1re intention qui comporte un hémogramme avec frottis sanguin pour rechercher une thrombopénie (isolée ou non) et préciser la morphologie plaquettaire, un bilan d’hémostase primaire avec temps d’occlusion plaquettaire (paralysie flasque aiguë [PFA]), et un dosage du facteur Willebrand (activité et antigène).
Remarque : la filière Maladies Rares MEHEMO fournit sur son site internet des informations claires et pratiques sur les pathologies hémorragiques.
En cas de thrombopénie associée (6, 7, 8, 9) :
(6) Le purpura thrombopénique immunologique (PTI) est un motif fréquent de consultation en pédiatrie. La démarche diagnostique et la prise en charge sont détaillées dans le Protocole national de diagnostic et de soins (PNDS).
(7) Les microangiopathies thrombotiques (MAT), principalement représentées en pédiatrie par le syndrome hémolytique et urémique (SHU), associent une anémie hémolytique mécanique avec présence de schizocytes, une thrombopénie de consommation, une insuffisance rénale avec protéinurie et parfois des signes d’ischémie multiviscérale. Le bilan doit comporter la recherche du facteur ADAMTS13 (activité et anticorps anti-ADAMTS13). La prise en charge doit être coordonnée avec un néphrologue référent.
(8) Les autres causes rares de thrombopénies périphériques peuvent être liées à un hypersplénisme dans le cadre d’une hépatopathie, ou bien une consommation comme dans la coagulation intravasculaire disséminée (CIVD). Voir Pas à pas Thrombopénies.
(9) Purpura sur thrombopénie centrale : l’analyse cyto logique du frottis médullaire (myélogramme) met en évidence une absence de mégacaryocyte. Ceci peut se retrouver dans deux cadres étiologiques :
a/ moelle envahie par des cellules tumorales (leucémie, lymphome, neuroblastome) ;
b/ hypoplasie ou aplasie médullaire : origine génétique (anémie de Fanconi et autres maladies génétiques rares), origine infectieuse (parvovirus B19, herpès virus), syndromes prétumoraux (myélodysplasie), origine idiopathique/auto-immune (qui est un diagnostic d’élimination, l’auto-immunité ne pouvant pas être mise en évidence, mais également la forme la plus fréquente d’aplasie chez l’enfant).
En cas d'autres anomalies de l’hémostase (10, 11) :
(10) La maladie de Willebrand est liée à un défaut génétique de concentration, de structure ou de fonction du facteur von Willebrand. Le diagnostic repose sur l’association de signes hémorragiques (épistaxis, ecchymoses, ménorragies, saignement prolongé pour blessure cutanée minime), l’existence d’antécédents familiaux et la diminution du taux de facteur von Willebrand. Un avis spécialisé est indispensable.
(11) Les thrombopathies congénitales sont rares. Le taux de plaquettes peut être normal ou diminué. Elles sont suspectées en présence d’antécédents familiaux de thrombopathie, d’anomalies morphologiques plaquettaires et d’une perturbation de l’hémostase primaire. Elles peuvent être associées à une thrombo pénie et à des signes extra-hématologiques (eczéma et déficit immunitaire dans le syndrome de Wiskott-Aldrich, anomalies du développement osseux ou intellectuel, insuffisance rénale, etc.).
Conclusion
L’évaluation précise d’un enfant présentant un purpura permettra d’orienter la prise en charge. Le diagnostic repose essentiellement sur l’interrogatoire et l’examen clinique. Des examens complémentaires simples et accessibles compléteront la démarche diagnostique. Il faut en priorité éliminer un purpura fulminans ou toute autre forme d’infection invasive à méningocoque. Hors contexte infectieux, le purpura rhumatoïde et le purpura thrombopénique immunologique sont à évoquer chez l’enfant.
Liens d’intérêts
Les auteurs déclarent ne pas avoir de liens d’intérêts avec cet article.
Références
Leung AK, Chan KW. Evaluating the child with purpura. Am Fam Physician 2001;64(3):419-28.
Perera TB, Murphy-Lavoie HM. Purpura Fulminans. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 [cité 3 avr. 2019]. disponible sur : https://www.ncbi.nlm.nih.gov/books/NBK532865/
Roberts PF, Waller TA, Brinker TM, Riffe IZ, Sayre JW, Bratton RL. Henoch-Schönlein Purpura: A Review Article: Southern Medical Journal 2007;100(8):821-4.
Joly B, Coppo P, Veyradier A. Le purpura thrombotique thrombocytopénique à révélation pédiatrique. Revue d’Oncologie Hématologie Pédiatrique 2017;5(3):111-9.
PNDS, Purpura thrombopénique immunologique de l’enfant et de l’adulte, sur www.has-sante.fr : https://www.has-sante.fr/portail/upload/docs/application/pdf/2017-06/dir36/pnds-_purpura_thrombopenique_immunologique.pdf