
Vous êtes ici
Thématique:
Ictère du nourrisson
P. Broué, O. Cuvinciuc
Gastroentérologie, Hépatologie et Nutrition Pédiatrique, Hôpital des enfants, avenue de Grande Bretagne, 31059 Toulouse Auteur correspondant. - Adresse e-mail : broue.p@chu-toulouse.fr (P. Broué)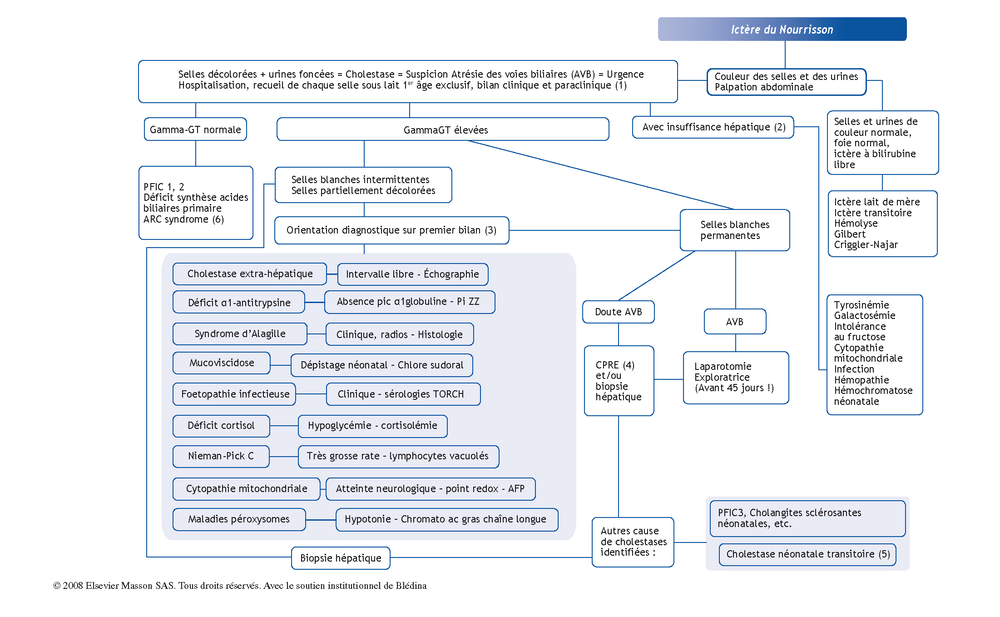
Arbre décisionnel - Commentaires
(1) Exploration initiale des cholestases du nourrisson :
-
suspicion d’AVB (atrésie des voies biliaires) jusqu’à preuve du contraire. La clinique est primordiale : examen soigneux et recueil de chaque selle en milieu hospitalier pendant 2 à 3 jours en supprimant l’absorption de tout élément pouvant éventuellement colorer les selles ;
-
pendant cette période d’observation, il faut réaliser les examens de base : Numération formule sanguine, bilan d’hémostase complet, bilan hépatique (ASAT, ALAT, GammaGT, Phosphatases alcalines, bilirubine totale et conjuguée), cholestérolémie, alpha-fœtoprotéine (AFP), électrophorèse des protides, chlore sudoral, examen ophtalmologique (fond d’œil et lampe à fente), radiographie du rachis. Echographie abdominale ;
-
l’échographie hépatique est habituellement sans particularité dans l’AVB sauf en cas de kyste biliaire sous-hépatique ou de syndrome de polysplénie. La présence ou l’absence de visualisation de vésicule est sans valeur.
(2) Insuffisance hépatique du nourrisson :
-
TP abaissé < 60 %, fibrinémie basse, le facteur V peut être normal longtemps ;
-
maladies métaboliques fréquentes. Intervalle libre variable après la naissance sauf cytopathie mitochondrie ;
-
infections plus rares : état de choc infectieux bactérien, herpes hominis type 6, herpes simplex 1 et 2, syphilis congénitale, autres virus… ;
-
lymphohistiocytose familiale et autres hémopathies ;
-
hémochromatose néonatale : hypotrophie, prématurité, insuffisance hépatique d’emblée, ferritinémie élevée, pathologie de l’allo immunisation fœto-maternelle.
(3) Autres cholestases du nourrisson à GGT élevées :
-
cholestase extra-hépatique du nourrisson :
-
diagnostic échographique ;
-
lithiase du cholédoque ;
-
rupture de la voie biliaire principale (formation liquidienne sous-hépatique) ;
-
sténose de la voie biliaire principale ;
-
kyste du cholédoque ;
-
-
syndrome d’Alagille : association variable d’un faciès particulier, cri aigu, atteinte cardio-vasculaire (sténose périphérique des artères pulmonaires = souffle systolique axillaire), embryotoxon postérieur, vertèbres dorsales en ailes de papillon et paucité des voies biliaires interlobulaires à l’histologie hépatique ;
-
les embryofœtopathies infectieuses associent une micro-céphalie, une importante hépatosplénomégalie et une hypo-trophie. Sérologies embryofœtopathies selon clinique. Attention, la primoinfection à cmV est banale et ne doit pas faire remettre en question à tort le diagnostic d’AVB ;
-
PFIC (Progressive Familal Intrahepatic Cholestasis) : cholestase fibrogène familiale. Classification en 3 types : types 1 et 2 (GammaGT normales) et type 3 (gammaGT élevées). Le diagnostic repose sur l’examen histologie du foie, la normalité des voies biliaires en cholangiographie, le dosage des acides biliaires et des phospholipides dans la bile ainsi que sur l’étude génétique ;
-
la cholangite sclérosante à début néonatal mime cliniquement l’atrésie biliaire. Histologiquement, il existe une paucité des voies biliaires interlobulaires. La cholangiographie est caractéristique ;
-
autres causes : Kabuki syndrome, Cholestase associée à la nutrition parentérale, fœtopathie alcoolique, déficit en citrine, déficit en transaldolase, septicémie, infection urinaire à E Coli, syndrome de Rotor, syndrome de Dubin-Johnson, CDG syndrome, maladie de gaucher, maladie de Wolman, trisomie, syndrome de Turner, Déficit en MRP2,…
(4) CPRE : Cholangiographie par cathétérisme rétrograde endoscopique de la papille.
(5) Cholestase idiopathique transitoire :cholestase de mécanisme inconnu associée à une souffrance néonatale dans la moitié des cas. Régression spontanée, durable, complète cli-nique et biologique avant la fin de la première année de vie. Attention, c’est un diagnostic d’élimination !
(6) ARC syndrome : syndrome associant arthrogrypose, anomalies rénales et cholestase.
Références
Bernard O. Diagnostic précoce des ictères cholestatiques chez le nouveau-né. Arch Pediatr 1998;5:1031-5.
Jacquemin E. Dépistage de l’atrésie des voies biliaires et couleur des selles : méthode de l’échelle colorimétrique. Arch Pediatr 2007;14:303-5.
Jacquemin E. Les cholestases néonatales : diagnostic et étiologie. Arch Pediatr 2001;8:412-4.