
Vous êtes ici
Thématique:
Docteur, est-ce un garçon ou une fille ?
C. Bouvattier1,*, D. Gorduza2
1Endocrinologie pédiatrique et centre de référence des anomalies du développement sexuel, hôpital Bicêtre, 78, rue du Général-Leclerc, 94275 Le Kremlin-Bicêtre Cedex, France2 Service de chirurgie pédiatrique et centre de référence des anomalies du développement sexuel, hôpital femme-mère-enfant, 59, boulevard Pinel, 69677 Bron Cedex, France Auteur correspondant - Adresse e-mail : claire.bouvattier@bct.aphp.fr (C. Bouvattier)
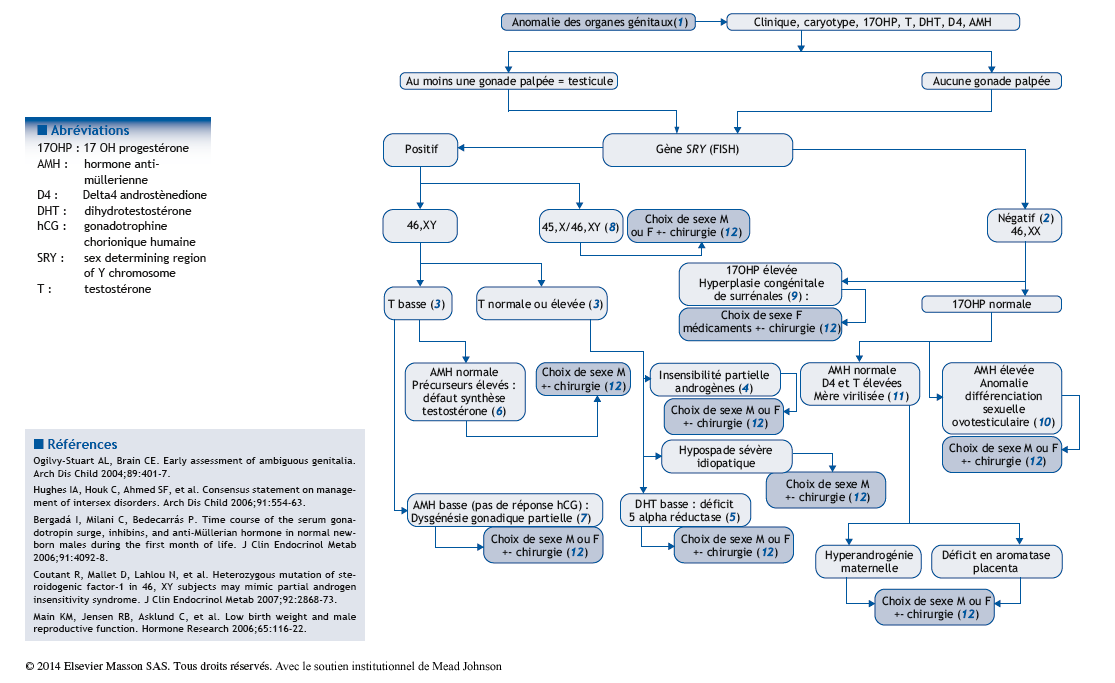
Arbre diagnostique – Commentaires
(1) La question se pose devant la découverte d’une anomalie des organes génitaux en salle de naissance. Celle-ci peut être un hypospade sévère chez un enfant ayant l’aspect d’un garçon, un gros clitoris, une cryptorchidie bilatérale avec testicules non palpés ou l’impossibilité de dire le sexe de l’enfant. Dans tous les cas, il faut être conscient du fait que, pour les parents, l’incapacité d’identifier son enfant dans le sexe masculin ou féminin est une situation très déstabilisante psychologiquement. On peut conseiller l’attribution d’un surnom au bébé. La découverte d’une anomalie des organes génitaux nécessite de surseoir transitoirement à la déclaration de l’enfant auprès des autorités d’état civil. L’article 288 du code d’état civil autorise les parents, si le médecin ne peut pas donner d’indication sur le sexe probable du nouveau-né, à demander au procureur de la République qu’aucune mention de sexe ne soit initialement inscrite dans l’acte de naissance. Une description anatomique précise est nécessaire, dans des termes indifférenciés (bourgeon génital, bourrelets génitaux, gonades). L’examen clinique doit être complet et cherchera une dysmorphie, des anomalies squelettiques, rénales, anales ou cutanées. Les examens doivent être orientés par les hypothèses diagnostiques. La présence du gène SRY par FISH ou PCR permet un diagnostic de sexe rapide. Le résultat du caryotype arrivera secondairement. Le dosage de la 17OH-progestérone (ne pas attendre le résultat du dépistage néonatal) est primordial, il dépiste les anomalies de la différenciation sexuelle en rapport avec des déficits enzymatiques dans lesquels une insuffisance surrénalienne peut engager le pronostic vital. Avant H36 de vie, des dosages de testostérone, AMH, inhibine B, D4 sont prescrits et une échographie pelvienne (présence ou non d’un utérus, malformation associée) réalisée. La question du choix de sexe est difficile. Il dépend de l’anatomie, des possibilités de la chirurgie, de ce qui est connu de l’évolution pubertaire… Nous connaissons très mal, chez l’homme, ce qui détermine l’identité sexuée d’une personne.
(2) La détection par FISH de SRY est la façon la plus rapide d’identifier la présence ou non d’un chromosome Y. Elle ne dispense pas d’un caryotype ultérieur.
(3) Le dosage de testostérone doit être réalisé dans les 36 premières heures de vie, ou après J15 de vie. Il permet d’évaluer la fonction leydigienne du testicule. Le dosage d’AMH, à n’importe quel moment, permet d’évaluer la fonction sertolienne du testicule.
(4) Une sécrétion normale ou élevée de testostérone chez un garçon porteur d’un hypospade doit faire évoquer le diagnostic d’insensibilité partielle à la testostérone (IPA). Il est probable que des événements (encore inconnus) altérant le développement du testicule au premier trimestre de la grossesse (sécrétion de testostérone retardée ?) peuvent donner, au moment de la naissance, une sécrétion et une sensibilité de la testostérone normales. Le diagnostic néonatal d’IPA est difficile. Un test de stimulation de la testostérone par l’hCG évalue 2 choses : la capacité du testicule à produire la testostérone et son action sur les organes cibles (augmentation de volume de la verge). Le récepteur des androgènes, porté par le chromosome X, peut être séquencé. De très nombreuses mutations sont décrites.
(5) Le déficit en 5alpha reductase de type II entraîne un défaut de virilisation souvent majeur des garçons, avec un phénotype néonatal très féminin à la naissance. Une virilisation pubertaire survient, au moment de l’augmentation d’expression de la 5alpha réductase de type I.
(6) Les défauts de la synthèse de la testostérone sont rares. Ils impliquent des déficits enzymatiques testiculaires isolés (déficit en 17bêta HSD), ou communs à la surrénale et la gonade (déficit en StAR, P450sc, P450c17, 3bêta HSD).
(7) Quand la testostérone et l’AMH sont basses, le diagnostic à évoquer est celui de défaut précoce de développement de la gonade (dysgénésies gonadiques). Il s’agit d’anomalies du passage de la gonade primordiale indifférenciée au testicule. Le développement des cellules de Leydig est insuffisant : la testostérone est basse, le développement des cellules de Sertoli a été altéré : l’AMH est basse. Parfois, des dérivés müllériens ont persisté (utérus, vagin).
(8) Des anomalies chromosomiques peuvent être associées à l’hypospade, comme la formule mosaïque 45, X/46, XY. Dans les dysgénésies gonadiques asymétriques 45, X/46, XY, cohabitent un testicule dysgénétique, une bandelette fibreuse et des dérivés müllériens unilatéraux.
(9) L’hyperplasie congénitale des surrénales (HCS) doit être éliminée chez un nourrisson aux organes génitaux anormaux, et sans gonade palpée. La 17OH progestérone, la testostérone et la rénine seront mesurées.
(10) Chez une fille 46, XX, une fois l’HCS éliminée, la cœlioscopie peut permettre le diagnostic d’anomalie de la différenciation sexuelle ovotesticulaire en affirmant dans l’abdomen la présence de tissu testiculaire et de tissu ovarien.
(11) Les causes de virilisation d’un fœtus féminin par sa mère sont exceptionnelles. Des signes d’hyperandrogénie sont présents chez la mère pendant la grossesse. Les signes de virilisation disparaissent chez le nouveau-né après la naissance.
(12) Le but de l’intervention chirurgicale est de redonner une anatomie et une fonctionnalité à l’appareil génital en réalisant une chirurgie de féminisation ou de masculinisation.
La chirurgie dite de « féminisation » : ses principes reposent sur la réduction de la taille du clitoris (clitoroplastie) si besoin, l’ouverture de la cavité vaginale au périnée ou la création d’une cavité vaginale si le vagin n’existe pas (vaginoplastie) et la plastie des lèvres et de la vulve (périnéoplastie). Si l’enfant présente une ou des gonades dysgénétiques ayant un potentiel de transformation maligne, elles seront retirées.
La chirurgie dite de « masculinisation » est essentiellement représentée par la chirurgie de la verge hypospade qui comporte trois étapes essentielles : le redressement du tubercule génital, la reconstruction de l’urètre manquant (urétroplastie) et la reconstruction de la face ventrale et du fourreau de la verge. Plusieurs techniques de reconstruction de l’urètre sont couramment pratiquées et leur choix repose sur la sévérité de l’hypospade.
À cette chirurgie du tubercule génital peut s’associer une chirurgie d’abaissement des testicules qui est très fréquente ou d’ablation des résidus müllériens.
Liens d’intérêts
Les auteurs ont déclaré n’avoir aucun conflit d’intérêts relatif à cet article.
Références
Ogilvy-Stuart AL, Brain CE. Early assessment of ambiguous genitalia. Arch Dis Child 2004;89:401-7.
Hughes IA, Houk C, Ahmed SF, et al. Consensus statement on management of intersex disorders. Arch Dis Child 2006;91:554-63.
Bergadá I, Milani C, Bedecarrás P. Time course of the serum gonadotropin surge, inhibins, and anti-Müllerian hormone in normal newborn males during the first month of life. J Clin Endocrinol Metab 2006;91:4092-8.
Coutant R, Mallet D, Lahlou N, et al. Heterozygous mutation of steroidogenic factor-1 in 46, XY subjects may mimic partial androgen insensitivity syndrome. J Clin Endocrinol Metab 2007;92:2868-73.
Main KM, Jensen RB, Asklund C, et al. Low birth weight and male reproductive function. Hormone Research 2006;65:116-22.