
Vous êtes ici
Thématique:
Démarche diagnostique devant une pneumopathie interstitielle
![]()
R. Epaud1,2,3,4,*, C. Delestrain1,2,3,4, E. Nattes1,2, N. Nathan3,5,6
1Service de pédiatrie, centre intercommunal de Créteil, Créteil, France 2Inserm, unité 955, équipe 5, Créteil, France 3Centre de référence des maladies respiratoires rares, Respirare®, Paris, France 4Université Paris-Est, faculté de Médecine, Créteil, France 5Service de pneumologie pédiatrique, AP-HP, hôpital Trousseau, Paris, France 6Inserm, UMR_S933, hôpital Trousseau, Paris, France Auteur correspondant - Adresse e-mail : ralph.epaud@chicreteil.fr (R. Epaud).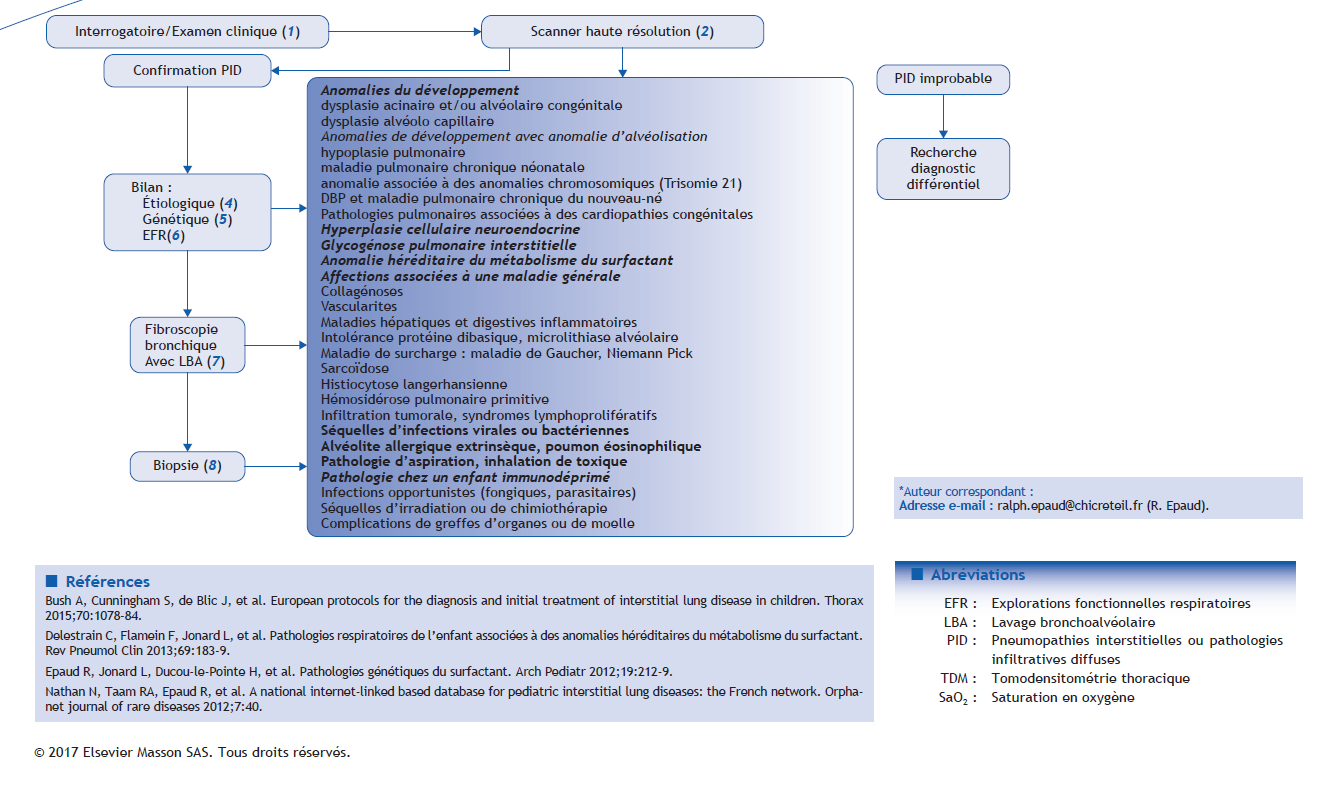
Cliquez sur l'image pour l'agrandir.
Arbre décisionnel – Commentaires
Les pneumopathies interstitielles ou pathologies infiltratives diffuses (PID) de l’enfant sont un groupe hétérogène de maladies respiratoires rares qui ont en commun une perturbation des échanges gazeux et des infiltrats pulmonaires diffus à l’imagerie. Les PID peuvent impliquer non seulement l’interstitium pulmonaire mais aussi les voies aériennes distales, les alvéoles, le lit vasculaire, les lymphatiques et l’espace pleural. Contrairement à l’adulte, peu de données épidémiologiques sont disponibles pour les PID de l’enfant. Une étude menée au Royaume-Uni et en Irlande entre 1995 et 1998 révèle une prévalence de 3,6 pour un million d’habitants chez les enfants de moins de 16 ans.
(1) Interrogatoire et examen clinique
Anamnèse
-
Antécédents (familiaux et personnels) ;
-
Origine ethnique ;
-
Consanguinité ;
-
Environnement ;
-
Prise de médicaments, antécédents d’irradiation ;
-
Hémoptysies ;
-
Antécédent de virose sévère (grippe, adénovirus).
Symptôme non spécifique
Polypnée, toux, dyspnée, intolérance à l’effort, infections respiratoires récidivantes, cassure de la courbe de poids.
Chez le nouveau-né
-
Détresse respiratoire sévère de début très précoce chez un enfant né à terme ;
-
Éliminer la cardiopathie ;
-
Présence d’infiltrats diffus à la radiographie du thorax.
Chez le nourrisson
-
Début plus progressif ;
-
Symptômes respiratoires : dyspnée au biberon ou à l’effort, toux sèche, wheezing, polypnée, tirage, cyanose ;
-
Symptômes extra-respiratoires : cassure de la courbe de poids, fièvre, hémoptysie.
Chez le plus grand enfant
-
Idem nourrisson mais installation plus lente ;
-
Dyspnée d’effort ou au repos, cyanose à l’effort ;
-
Insuffisance respiratoire chronique avec hypoxie et hippocratisme digital.
Examen clinique
Examen pulmonaire
-
Cyanose, Hypoxémie (SaO2 < 95 %), hippocratisme digital ;
-
Fréquence respiratoire, signes de lutte, wheezing ;
-
Auscultation : crépitants, sibilants, signes de condensation pulmonaire.
Recherche de signes extra-pulmonaires orientant vers une étiologie particulière
-
Lésion cutanée ;
-
Trouble neurologique ;
-
Hépato-splénomégalie ;
-
Adénopathies périphériques ;
-
Hypertension artérielle pulmonaire.
(2) Scanner thoracique haute résolution
La tomodensitométrie thoracique (TDM) sera réalisée dans un centre spécialisé en imagerie pédiatrique. La rapidité de la technique permet, le plus souvent, d’éviter sa réalisation sous sédation ou anesthésie générale, après la prise du biberon chez le nourrisson et/ou avec une simple contention chez les enfants de moins de 4-5 ans. Avant l’âge de 5-6 ans, elle sera réalisée en respiration calme. Après, une apnée en inspiration bloquée sera demandée à l’enfant. Ces acquisitions sont réalisées en mode volumétrique avec reconstructions de 1 à 1,25 mm chevauchées avec algorithme en haute résolution pour l’étude du parenchyme pulmonaire. Le TDM peut être normal mais montre le plus souvent une infiltration diffuse avec des caractéristiques (images kystiques, nodules) qui peuvent orienter le diagnostic étiologique.
(4) Bilan étiologique
Examens biologiques
-
NFS (numération et formule sanguine), VS (vitesse de sédimentation), CRP, ionogramme sang et urine ;
-
Recherche d’hématurie, de protéinurie ;
-
Biologie hépatique ; • Intradermoréaction à la tuberculine (IDR) ou quantiferon ;
-
Enzyme de conversion de l’angiotensine, Bilan phosphocalcique, Calcitriol, lysozyme ;
-
Ammoniémie, chromatographie des acides aminés ;
-
Bilan immunitaire (étude quantitative et fonctionnelles des lymphocytes, polynucléaire et macrophage ;
-
Sérologies (virale, précipitines aviaires) ;
-
Bilan d’auto-immunité.
Échocardiographie
Examen Ophtalmologique (lampe à fente)
Radiographie du squelette (histiocytose langerhansienne ?)
Biopsie : glandes salivaires accessoires et biopsie cutanée si lésion, capillaroscopie si auto-immunité
pHmétrie : si suspicion de pathologie d’inhalation
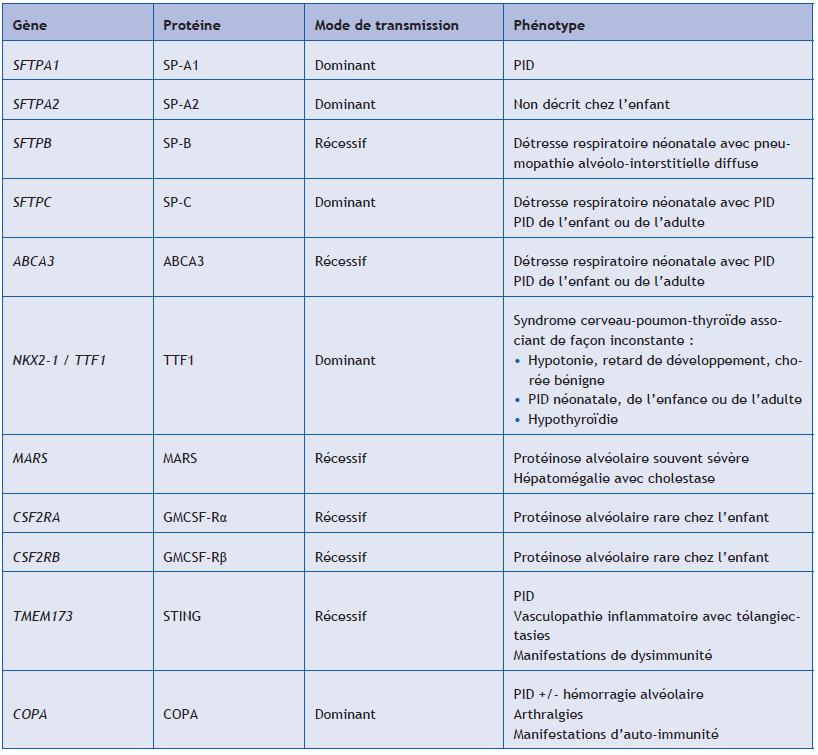
(5) Étude génétique
Une cause génétique est identifiée à ce jour chez environ 20 % des patients présentant une PID. Une analyse génétique est recommandée pour tous les patients pédiatriques qui présentent une PID chronique non étiquetée, qu’elle soit sporadique ou familiale. L’interprétation des résultats est réalisée en fonction du phénotype du patient, du caractère éventuellement familial de la PID et de la notion d’une éventuelle consanguinité parentale. L’analyse doit être réalisée par un laboratoire de génétique spécialisé, et la mise en évidence d’une anomalie génétique doit toujours s’accompagner, pour le patient et sa famille, d’une consultation de conseil génétique.
(6) Exploration fonctionnelle respiratoire/gaz du sang
Chez le nourrisson et l’enfant, permet comme chez l’adulte une analyse physiopathologique de la pathologie respiratoire, le suivi de la croissance pulmonaire et l’efficacité thérapeutique. Avant l’âge de 3 ans, l’exploration ne peut être réalisée que pendant le sommeil et nécessite donc l’utilisation d’une prémédication. Entre 3 et 6-8 ans, l’exploration demande une coopération active (accepter au moins un pince-nez et un embout buccal). Après l’âge de 6-8 ans, l’exploration se rapproche de celle de l’adulte. La première exploration fonctionnelle respiratoire (EFR) devrait se situer dans le délai le plus court possible après que le diagnostic a été posé si l’état de l’enfant le permet. La fonction respiratoire est évaluée par la mesure des volumes pulmonaires (capacité pulmonaire totale, capacité vitale, capacité résiduelle fonctionnelle) et la mesure de la diffusion qui selon l’âge sera réalisée par la technique en rebreathing ou en apnée. Les mesures qui permettent aussi d’éliminer un syndrome obstructif doivent être également réalisées (résistances, volumes et débits expiratoires forcés). L’hypoxémie (PaO2 < 80 mm Hg) est recherchée par un gaz du sang mais l’atteinte des échanges gazeux n’est observée que dans les formes sévères et la réalisation d’une épreuve d’effort peut démasquer une hypoxémie dans des atteintes moins importantes. L’épreuve de marche de 6 minutes est particulièrement adaptée dans ce type de pathologie et peut être réalisée à partir de l’âge de 4 ans. L’évaluation des besoins en oxygène dans la journée et la nuit ne nécessitent pas la réalisation d’une polysomnographie. Une évaluation simple de la saturation diurne et nocturne peut être suffisante pour aider à la prise en charge des patients.
Fibroscopie et lavage broncho-alvéolaire
La fibroscopie est réalisée au mieux sous anesthésie générale. Un lavage broncho-alvéolaire est effectué avec étude cytologique, colorations de base (May-Grünwald Giemsa [MGG], Papanicolaou, Perls) et colorations complémentaires (Ziehl, Grocott, PAS [periodic acid schiff]). Le reste du liquide est centrifugé, le surnageant est cryopréservé et le culot cellulaire conservé à – 80 °C.
(7) Le profil du LBA permettra d’orienter le diagnostic :
-
Sarcoïdose : cellularité augmentée, augmentation des lymphocytes T CD4+.
-
Pneumopathie d’hypersensibilité : cellularité augmentée, augmentation des lymphocytes T CD8+, des lymphocytes à grains azurophiles, des polynucléaires neutrophiles et éosinophiles.
-
Tuberculose : cellularité augmentée, augmentation des lymphocytes T CD4+, présence de bacilles acide-alcoolo-résistants (BAAR) sur la coloration de Ziehl.
-
Protéinose alvéolaire : aspect laiteux du LBA avec accumulation de matériel lipoprotéique PAS+ avec des macrophages au cytoplasme très abondant microvacuolisé.
-
Histiocytose Langerhansienne : cellularité augmentée avec augmentation des macrophages, des lymphocytes, des polynucléaires éosinophiles. Les histiocytes Langerhansiens sont identifiés par le CD1a et la langérine.
-
Fibrose : cellularité augmentée, augmentation des polynucléaires neutrophiles.
-
Hémorragie alvéolaire et hémosidérose : taux et score de Golde augmentés sur la coloration de Perls.
(8) Biopsie pulmonaire
La biopsie est réalisée par ponction transbronchique, thoracoscopie ou thoracotomie. Les prélèvements sont fixés dans le formol (microscopie optique), le glutaraldéhyde (microscopie électronique) et cryopréservés. L’examen au microscope est réalisé sur les colorations standards (HES [hématoxine éosine safran]), les colorations spéciales (Perls, PAS, Grocott, réticuline, trichrome de Masson) et les immunomarquages (TTF-1, bombésine, protéines du surfactant, marqueurs vasculaires CD31, CD34, D2-40).
Liens d’intérêts
Les auteurs n’ont pas transmis leurs liens d’intérêts.
Références
Bush A, Cunningham S, de Blic J, et al. European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax 2015;70:1078-84.
Delestrain C, Flamein F, Jonard L, et al. Pathologies respiratoires de l’enfant associées à des anomalies héréditaires du métabolisme du surfactant. Rev Pneumol Clin 2013;69:183-9.
Epaud R, Jonard L, Ducou-le-Pointe H, et al. Pathologies génétiques du surfactant. Arch Pediatr 2012;19:212-9.
Nathan N, Taam RA, Epaud R, et al. A national internet-linked based database for pediatric interstitial lung diseases: the French network. Orphanet journal of rare diseases 2012;7:40